Definiciones y etiopatogeniaArriba
El síndrome de Cushing es un conjunto de síntomas y signos clínicos secundarios al exceso de glucocorticoides. El hipercortisolismo subclínico se suele diagnosticar en el transcurso de las pruebas realizadas para el estudio de un incidentaloma suprarrenal. Está causado por un incremento leve de los niveles de glucocorticoides provocado por una secreción excesiva de cortisol por un tumor suprarrenal, que inhibe la secreción de glucocorticoides por la glándula suprarrenal contralateral. En este caso no aparecen los síntomas característicos, a pesar de que puedan desarrollarse con mayor frecuencia diabetes mellitus, obesidad abdominal, hipertensión arterial, eventos cardiovasculares y osteoporosis.
Clasificación etiológica del síndrome de Cushing:
1. Síndrome de Cushing endógeno: ocurre a consecuencia de una secreción excesiva de glucocorticoides suprarrenales
1) Síndrome de Cushing independiente de ACTH (hipercortisolismo primario)
a) Tumores suprarrenales autónomos, que en general son solitarios y con menor frecuencia adenomas múltiples; cáncer suprarrenal →Carcinoma suprarrenal. Los tumores que se originan en las células de la zona fasciculada de la CS secretan de manera excesiva solo cortisol, mientras que los otros tipos de tumores (de la zona reticular o tumores mixtos) secretan también andrógenos. El exceso de cortisol inhibe la secreción de CRH y de ACTH → se produce atrofia del tejido corticosuprarrenal contralateral e ipsilateral localizado fuera del tumor. A menudo se encuentran múltiples nódulos en la CS, denominados hiperplasia nodular, que tienen su origen en cambios policlonales, a diferencia de los adenomas, que son debidos a proliferación monoclonal.
b) Hiperplasia macronodular suprarrenal: está causada por la presencia en la corteza suprarrenal de receptores ectópicos, que responden a estímulos atípicos, principalmente al péptido inhibidor gástrico (GIP, su nombre actual: péptido insulinotrópico dependiente de la glucosa), de secreción posprandial en el tubo digestivo. Otros factores estimulantes son: catecolaminas, vasopresina, TSH, hormona luteinizante, gonadotropina coriónica humana (hCG), FSH, estrógenos en concentraciones altas, PRL e interleucina-1.
c) Hiperplasia suprarrenal micronodular (hiperplasia [displasia] primaria suprarrenal micronodular pigmentada): hay una variante familiar condicionada genéticamente, denominada síndrome de Carney, que aparece junto con otras anomalías como los mixomas cutáneos, cardíacos, mamarios, lentiginosis cutánea, tumores testiculares, y a veces otros trastornos endocrinos (p. ej. acromegalia), y una variante esporádica en la cual pueden jugar un papel las inmunoglobulinas que estimulan la hipertrofia de la corteza suprarrenal. Al igual que en otras variantes independientes de ACTH, el tejido suprarrenal fuera de los nódulos puede estar atrofiado.
2) Síndrome de Cushing dependiente de ACTH (hipercortisolismo secundario): variante hipofisaria (secundario a la excesiva producción de ACTH por un tumor hipofisario, es decir enfermedad de Cushing →Enfermedad de Cushing; es la causa más frecuente del síndrome de Cushing), secreción ectópica de ACTH por un tumor no hipofisario (causa mucho menos frecuente) o síndrome de la secreción de CRH ectópica (la menos frecuente, p. ej. carcinoma pulmonar microcítico, neoplasias neuroendocrinas).
2. Síndrome de Cushing exógeno: causado por la administración de glucocorticoides a dosis mayores que las sustitutivas (la causa más frecuente de síndrome de Cushing), independientemente de la forma de administración del fármaco (comprimidos, preparados inhalados, ungüentos, inyecciones, incluidas las intraarticulares).
Cuadro clínico e historia naturalArriba
1. Síntomas: cambios del rostro o de la silueta, debilidad muscular y baja tolerancia al esfuerzo físico, susceptibilidad de la piel a traumatismos (ulceraciones difíciles de curar, hematomas frecuentes); polidipsia y poliuria (→ controlar la glucemia; en casos graves puede desarrollarse un síndrome hiperglucémico hiperosmolar); hiperfagia; cefalea y vértigo (→ controlar la presión arterial); labilidad emocional, tendencia a la depresión, empeoramiento de la memoria, rara vez estados psicóticos; dolores óseos (en caso de osteoporosis → se deben buscar fracturas patológicas de los cuerpos vertebrales, costillas y huesos púbicos e isquiones); tendencia a infecciones, especialmente infecciones oportunistas (p. ej. fúngicas), a menudo de curso grave, también a la tuberculosis; síntomas de la enfermedad isquémica del corazón (→ control del lipidograma), de insuficiencia cardíaca o enfermedad tromboembólica venosa (efecto protrombótico de glucocorticoides), síntomas de enfermedad ulcerosa gástrica o duodenal (especialmente en pacientes que ingieren AINE); síntomas de litiasis (a causa de hipercalciuria e hiperfosfaturia); disminución de la libido en hombres, en mujeres oligomenorrea o amenorrea secundaria.
2. Signos: obesidad central con unos característicos depósitos del tejido adiposo de la cara, la región interescapular (“joroba de búfalo”) y el tronco, con almohadillas de grasa en las fosas supraclaviculares y con las extremidades delgadas; cara redonda (cara de luna llena), a menudo roja (a causa de la policitemia y del adelgazamiento de la piel), con vasodilatación; cuello corto con depósitos de grasa; atrofia muscular en las extremidades y el tronco; estrías anchas rojas o purpúreas en la piel del abdomen, de las caderas, mamas, muslos y en pacientes jóvenes también en las fosas axilares, cubitales y hueco poplíteo (→ diferenciarlo de las estrías estrechas, de color rosa que aparecen en jóvenes, asociadas a un rápido aumento de peso y que palidecen con el tiempo); adelgazamiento de la piel, tendencia a la aparición de equimosis en la piel, a veces petequias espontáneas; signos de hiperandrogenismo de diferente grado de expresión: acné e hirsutismo (→ diagnóstico diferencial con el síndrome de ovario poliquístico); hipertensión arterial (en la mayoría de los enfermos, en general leve o moderada), hiperpigmentación de la piel (aparecerá en aquellos enfermos en los que se mantiene durante largo tiempo una concentración alta de ACTH); edema de los miembros inferiores.
3. Historia natural: en el síndrome de Cushing subclínico, incluso de larga duración, puede no llegarse a desarrollar el cuadro clínico típico; el riesgo de progresión hasta el síndrome de Cushing clínico no es alto, por lo que en general no se debe tratar el síndrome de Cushing subclínico como una fase temprana del síndrome de Cushing. El síndrome de Cushing clínico suele verse en un estadio avanzado de una enfermedad de curso largo. Con una frecuencia mucho mayor aparecen solo algunos síntomas, p. ej. intolerancia a la glucosa o diabetes, dislipidemia, hipertensión arterial y aumento rápido del peso corporal (obesidad); también aumenta el riesgo de la osteoporosis.
DiagnósticoArriba
El síndrome de Cushing se debe sospechar y diagnosticar en las siguientes situaciones:
1) en enfermos con manifestaciones múltiples de hipercortisolismo y de rápida progresión, especialmente ante la presencia de los signos más característicos (estrías típicas, atrofia de músculos proximales de las extremidades inferiores y cintura escapular, eritema facial, hematomas espontáneos)
2) en enfermos con un curso atípico de hipertensión arterial, diabetes mellitus u osteoporosis, que puede sugerir formas secundarias (especialmente en caso de resistencia al tratamiento y en personas jóvenes)
3) en caso de diagnosticar accidentalmente un tumor suprarrenal (incidentaloma).
El diagnóstico del síndrome de Cushing es difícil. No existe una prueba de tamizaje única que lo confirme o excluya. Al interpretar los resultados, hay que tener en consideración el cuadro clínico, así como las ventajas y desventajas de las pruebas específicas. El resultado positivo de una sola prueba de tamizaje no permite el diagnóstico seguro del síndrome de Cushing y es necesario realizar una prueba de confirmación. De la misma manera, en un paciente con características clínicas que sugieran hipercortisolismo, el resultado negativo de una sola prueba no es suficiente para la exclusión de forma segura del síndrome de Cushing. En cualquier caso siempre hay que excluir la administración reciente de glucocorticoides (síndrome de Cushing exógeno).
Exploraciones complementarias
1. Pruebas bioquímicas básicas: hipopotasemia e hiperpotasiuria, hipocalcemia e hipercalciuria, así como hipofosfatemia con hiperfosfaturia debido a su resorción deficiente en los túbulos renales; hiperglucemia (intolerancia a la glucosa o diabetes), aumento de las concentraciones del colesterol total, C-LDL y triglicéridos, disminución de la concentración de C-HDL.
2. Hemograma: eritrocitosis, leucocitosis y trombocitosis, concentración elevada de la hemoglobina, disminución del recuento de los linfocitos, eosinófilos y monocitos (debido a la migración hacia el espacio extravascular).
3. Pruebas hormonales del sistema hipotalámico-hipofisario-adrenal (los rangos de referencia pueden ser diferentes, hay que averiguar los rangos de referencia del laboratorio en el que se realizaron las pruebas):
1) Confirmación de la hipercortisolemia: pruebas en caso de sospecha del síndrome de Cushing →fig. 11.2-1
a) Ausencia del ritmo circadiano de la secreción de cortisol: aumento de la concentración nocturna (a las 23:00-24:00 h) del cortisol en el suero >149 mmol/l (5,4 μg/dl) o en la saliva (>4,0 nmol/l [145 ng/dl]). La concentración del cortisol por la mañana a menudo está dentro del rango de los valores de referencia.
b) Aumento de la eliminación del cortisol libre en la orina (para la exclusión se necesitan 2-3 colecciones de orina de 24 h): el resultado excede el LSN en 3-4 veces (330 nmol/24 h [120 µg/24 h]).
c) Disminución insuficiente de la concentración de cortisol en el test de supresión con 1 mg de dexametasona (prueba nocturna de supresión con dexametasona, test breve de supresión con dexametasona): indicar al paciente que ingiera 1 mg de dexametasona VO antes de dormir (a las 22:00-24:00). Evaluar la concentración sérica de cortisol por la mañana en ayunas, entre las 8:00 y las 9:00. En el test de dos días de supresión con 2 mg de dexametasona se administran al paciente 0,5 mg de dexametasona VO cada 6 h durante 2 días, y se determina la cortisolemia 48 h después de la primera dosis. Una concentración de cortisol <50 nmol/l (1,8 µg/dl) descarta el síndrome de Cushing con elevada fiabilidad. Recordar que un solo resultado anormal del test con dexametasona no puede constituir el fundamento de la decisión del tratamiento quirúrgico.
2) Evaluación de la causa de hipercortisolemia: pruebas de confirmación del síndrome de Cushing y de identificación de su etiología →fig. 11.2-2.
a) Nivel sérico de ACTH: depende de la etiología del síndrome de Cushing. Una concentración de ACTH de <2 pmol/l (10 ng/l) en paciente con hipercortisolemia es indicativo de la presencia de un síndrome de Cushing independiente de ACTH y de >4 pmol/l (20 ng/l) indica un síndrome de Cushing dependiente de ACTH. En el caso de concentración de 2-4 pmol/l (10-20 ng/l) hay que realizar la prueba de estimulación con CRH.
b) Prueba de estimulación con CRH (prueba de estimulación directa de secreción de ACTH e indirecta del cortisol por CRH): en la enfermedad de Cushing es característico el aumento de la concentración de ACTH de varias veces su valor normal tras la estimulación con CRH, pero en caso de valores iniciales visiblemente elevados se considera significativo el aumento de la concentración de ACTH de ≥35-50 % y del cortisol de ≥14-20 %. En el síndrome de Cushing independiente de ACTH en general no hay respuesta a la CRH o es mínima.
c) Prueba de supresión con 8 mg de dexametasona (2 mg cada 6 h por 2 días; actualmente se realiza raramente): los resultados dependen de la etiología del síndrome de Cushing; su papel consiste en diferenciar entre la enfermedad de Cushing y las variantes de la hiperfunción suprarrenal con exceso del cortisol (tumor de corteza suprarrenal funcionante, síndrome de ACTH ectópico e hiperplasia suprarrenal nodular). En general en la enfermedad de Cushing la eliminación de cortisol y de sus metabolitos disminuye en ≥50 % y en caso de la secreción autónoma de cortisol de causa adrenal o (con menor frecuencia) ectópica la supresión no ocurre. En la hiperplasia suprarrenal micronodular es característico un aumento de la eliminación diaria de cortisol cada vez más grande y un aumento gradual de la concentración de cortisol en sangre en los días posteriores a la prueba. El resultado de esta prueba no es fiable p. ej. en caso de presencia de un tumor ectópico que secreta ACTH y tiene expresión de los receptores de glucocorticoides.
d) Hiperreactividad suprarrenal a los estímulos atípicos: averiguar en caso de sospechar hiperplasia suprarrenal macronodular; medir la concentración sérica del cortisol inicial y a los 30, 60, 90 y 120 min después del desayuno o tras el test de tolerancia oral a la glucosa con 75 g; tras la verticalización; tras la ingesta de 10 mg de metoclopramida VO; tras la inyección iv. de 100 µg de GnRH o 200 µg de TRH. Un aumento de la concentración del cortisol confirma el diagnóstico.
PERSPECTIVA LATINOAMERICANA
El CRH no está disponible en Argentina.
4. Pruebas de imagen.
1) RMN de la hipófisis →Tumores hipofisarios.
2) TC o RMN suprarrenal: los hallazgos dependerán de la causa del síndrome de Cushing →fig. 11.2-1
a) Tumor/tumores autónomos corticosuprarrenales: en la TC se ve un tumor unilateral con rasgos de adenoma →Tumor suprarrenal detectado de forma accidental (incidentaloma); posibles rasgos de atrofia corticosuprarrenal contralateral e ipsilateral (con excepción del tumor). Más raramente los adenomas múltiples son bilaterales. En la RMN se observa un alto contenido de lípidos, un rápido lavado (washout) del contraste. Cáncer corticosuprarrenal funcionante →Carcinoma suprarrenal.
b) Hiperplasia suprarrenal macronodular: en la TC las glándulas suprarrenales aparecen simétricas, en general están aumentadas de tamaño, a menudo con un borde policíclico y con la misma densidad que la de los adenomas. En la RMN se ve un alto contenido suprarrenal de lípidos.
c) Hiperplasia suprarrenal micronodular: en la TC y RMN las glándulas suprarrenales son generalmente simétricas, de tamaño normal o ligeramente aumentadas de tamaño. El diagnóstico se establece durante la cirugía (es característica la coloración negra y amarilla, resultante de la presencia de lipofuscina en los nódulos).
3) Radiografía ósea: fracturas patológicas, focos de osteonecrosis, a menudo edad ósea retrasada en niños y jóvenes.
4) Densitometría ósea: disminución de la densidad mineral ósea (DMO), presencia de criterios de osteopenia u osteoporosis, especialmente en vértebras lumbares y en el extremo proximal del fémur (en el síndrome de Cushing, el riesgo de fracturas es mucho más elevado respecto al grado de disminución de la DMO).
5) Gammagrafía de receptores con análogos de somatostatina marcados para detectar neoplasias neuroendocrinas que secretan ACTH ectópico o gammagrafía con 131I-norcolesterol (análogo del colesterol marcado con un radioisótopo de yodo) dirigida a detectar un tumor adrenal de ubicación ectópica o a identificar un tumor autónomo en enfermos con tumores en ambas glándulas suprarrenales.
Criterios diagnósticos
Algoritmo diagnóstico →fig. 11.2-1 y fig. 11.2-2. Tanto la confirmación, como la exclusión del hipercortisolismo exige la realización de ≥2 pruebas diagnósticas distintas.
1. Síndrome de Cushing clínico: síntomas y signos del síndrome de Cushing, hipercortisolemia (inhibición incorrecta de la secreción de cortisol con 1 mg de dexametasona, aumento de la concentración nocturna del cortisol en el suero o en la saliva y/o aumento de la eliminación del cortisol libre en la orina) con una concentración plasmática de ACTH disminuida (<2 pmol/l [10 ng/l]; hipercortisolismo suprarrenal: síndrome de Cushing independiente de ACTH) o elevada (adenoma hipofisario o secreción ectópica de ACTH o CRH: síndrome de Cushing dependiente de ACTH); la respuesta en test con CRH depende de la causa de hipercortisolemia; falta de supresión de secreción del cortisol (en caso de tumor suprarrenal) o supresión solo por una dosis alta [8 mg] de dexametasona (adenoma hipofisario). En ocasiones elevación de los niveles de andrógenos (secretados por el tumor adrenal) y, en caso de un adenoma de la zona fascicular una disminución en los niveles de DHEA-S en el suero a causa de la deficiencia de ACTH. Tumor o tumores suprarrenales detectados en TC/RMN o tumor hipofisario en RMN o mucho menos frecuentemente: fuente ectópica de ACTH o CRH detectada en la gammagrafía de receptores.
2. Hipercortisolismo subclínico causado por un tumor autónomo o por tumores corticosuprarrenales (independiente de ACTH): establecer el diagnóstico según las mismas reglas que en la sospecha del síndrome de Cushing (→más arriba). No obstante, el manejo en caso de diagnosticarlo puede ser distinto: es controvertido el tratamiento quirúrgico. Al valorar las indicaciones para la cirugía, hay que tomar en cuenta el cuadro clínico (incluir las posibles consecuencias de la hipercortisolemia: osteopenia/osteoporosis, obesidad abdominal, hipertensión arterial, diabetes mellitus), así como los resultados de las siguientes exploraciones complementarias (para el diagnóstico se sugiere el cumplimiento de ≥2 criterios):
1) test con 1 mg de dexametasona: concentración sérica de cortisol ≥50 nmol/l (1,8 μg/dl)
2) concentración sérica matinal de ACTH <2 pmol/l (10 pg/ml)
3) eliminación diaria de cortisol libre en la orina >LSN
4) concentración sérica de cortisol a las 23:00-24:00 ≥149 nmol/l (5,4 μg/dl).
Diagnóstico diferencial
Diagnóstico diferencial entre el síndrome de Cushing dependiente de ACTH e independiente de ACTH →fig. 11.2-2.
Otras causas del exceso de glucocorticoides
1) Síndrome de resistencia a glucocorticoides: es un síndrome debido a la deficiencia parcial de sensibilidad del receptor para glucocorticoides (condicionado genéticamente, raro); aumento de la concentración sérica de ACTH, cortisol, andrógenos y aldosterona, pero con ausencia de síntomas del exceso del cortisol, síntomas de androgenización en mujeres y de hiperaldosteronismo. El ritmo circadiano de la secreción del cortisol está preservado tanto como la respuesta de la hipófisis y los suprarrenales a CRH. Tratamiento: dexametasona 1,0-1,5 mg/d con el fin de suprimir la secreción de ACTH.
2) Exceso de glucocorticoides funcional (el denominado síndrome pseudo-Cushing) en el que la hipercortisolemia no es un efecto de los cambios estructurales del sistema hipofisario-suprarrenal, sino de otros trastornos (la hipercortisolemia no requiere tratamiento)
a) depresión: hipercortisolemia y deficiencia de la supresión por la dexametasona, pero con el ritmo circadiano de cortisol preservado y la concentración de ACTH normal
b) embarazo: aumento de la concentración de transcortina (CBG) en la sangre y al mismo tiempo de cortisol total, determinado mediante las pruebas disponibles; también aumenta la eliminación de cortisol libre por la orina; la supresión por dexametasona a menudo está alterada, pero se conserva el ritmo circadiano de secreción de cortisol
c) alcoholismo: en pocos casos aparecen rasgos somáticos del síndrome de Cushing (cambio de metabolismo hepático del cortisol e influencia del alcohol sobre el SNC); la abstinencia lleva a la remisión de los síntomas
d) anorexia nerviosa: aumento de la concentración de cortisol, debido principalmente a la disminución del aclaramiento renal, pero también es posible un aumento de la secreción de ACTH. La deficiencia de supresión por la dexametasona es una manifestación de la resistencia adquirida del receptor para glucocorticoides, lo que explica la ausencia de hallazgos clínicos de la hiperfunción corticosuprarrenal.
TratamientoArriba
Tratamiento sintomático
Tratamiento de las complicaciones del síndrome de Cushing: de la hipertensión arterial, siendo los fármacos de elección los inhibidores de la convertasa de angiotensina o los bloqueantes del receptor de angiotensina, combinados con un calcioantagonista o antagonista del receptor de mineralocorticoides (espironolactona, eplerenona); a continuación se pueden pautar diuréticos, α-bloqueantes y β-bloqueantes. También se tratan los trastornos del metabolismo de carbohidratos y lípidos, osteoporosis y trastornos psíquicos. Algunas complicaciones remiten tras el tratamiento causal eficaz del síndrome de Cushing.
Tratamiento de la hipercortisolemia
Depende de la etiología del síndrome de Cushing. Es obligatorio en el síndrome de Cushing clínico y en caso de complicaciones de hipercortisolemia. El tratamiento de elección es la intervención quirúrgica, que depende de la etiología del síndrome de Cushing (la resección de adenoma en la enfermedad de Cushing, tumor suprarrenal o, con mucha menor frecuencia, el tejido neoplásico que provoca secreción ectópica de ACTH). Sin embargo, no se debe iniciar la terapia si el diagnóstico del síndrome de Cushing no es seguro. Según las recomendaciones de la Endocrine Society (2015), no debe iniciarse la terapia si las exploraciones hormonales arrojan valores limítrofes, o si no hay sintomatología característica del síndrome de Cushing.
1. Adenoma hipofisario →Enfermedad de Cushing.
2. Tumor/tumores autónomo(s) de la corteza suprarrenal: el método de elección es la resección quirúrgica del tumor suprarrenal tras la preparación preoperatoria con un inhibidor de la esteroidogénesis → administrar ketoconazol 400-1200 mg/d en 2-3 dosis divididas o metirapona 750-4000 mg/d en 3-4 dosis divididas. Inicialmente se obtiene la remisión bioquímica y, transcurridas ~3 semanas, aparecerá la mejoría clínica. Se debe tener cuidado en evitar la deficiencia de glucocorticoides, vigilar la aparición de síntomas de una crisis suprarrenal inminente (algunos tumores tienen una sensibilidad muy grande a los inhibidores de la síntesis de cortisol). El único inhibidor de la esteroidogénesis apto para la administración iv. es el etomidato. En los enfermos con enfermedad de Cushing que no se puedan intervenir, o cuya operación no haya tenido éxito, se puede administrar pasireotida, un análogo de la somatostatina de 2ª generación. Pretratamiento con glucocorticoides en caso de cirugía: igual que en enfermos con insuficiencia corticosuprarrenal →más arriba. El primer criterio de remisión de la insuficiencia corticosuprarrenal primaria tras el tratamiento quirúrgico es una concentración de cortisol matinal <138 nmol/l (5 µg/dl), o una excreción de cortisol libre por la orina <28-56 nmol/d (10-12 µg/d) hasta 7 días tras la resección selectiva del tumor suprarrenal (en dicho momento la liberación de ACTH está disminuida debido a la hipercortisolemia previa). Por esta razón, en el período posoperatorio se presenta un déficit de hormonas corticosuprarrenales que se prolonga desde varios meses hasta incluso 2 años y que requiere tratamiento sustitutivo con hidrocortisona. La insuficiencia corticosuprarrenal aparece también tras la resección de adenoma hipofisario corticotropo (que produce ACTH en exceso): la cirugía fue exitosa si en las primeras 24 h desde la misma la concentración de cortisol disminuye <70 nmol/l (<2,5 µg/dl).
3. Hiperplasia macronodular y micronodular suprarrenal: el método de elección es la resección bilateral de las glándulas suprarrenales; administración de hidrocortisona como en caso de la cirugía de adenoma.
PERSPECTIVA LATINOAMERICANA
La metirapona no está disponible en la Argentina, y el uso de ketoconazol es fuera de ficha técnica.
PronósticoArriba
En casos del síndrome de Cushing de larga duración (independientemente de la etiología) pueden aparecer complicaciones vasculares dependientes de la hipertensión arterial. Incluso en su forma leve, el síndrome de Cushing no tratado aumenta la mortalidad en 4 veces (principalmente debido a las enfermedades cardiovasculares e infecciones) en comparación con la población general. Tras un tratamiento quirúrgico eficaz, muchos síntomas del síndrome de Cushing, incluida la hipertensión arterial y la diabetes mellitus, ceden o disminuyen a lo largo de 12 meses. Durante ~5 años se mantiene un incremento en el riesgo de muerte por enfermedades cardiovasculares.
1. La resección de adenoma/adenomas corticosuprarrenales lleva a la remisión completa de los síntomas del síndrome de Cushing. La reaparición del tumor puede producirse por recidiva a nivel de los fragmentos de la corteza suprarrenal en el tejido graso. Sin embargo, no se produce recidiva del adenoma extirpado completamente. Puede ser necesario la administración de forma periódica del tratamiento de sustitución.
2. La adrenalectomía bilateral en caso de la hiperplasia macronodular o micronodular lleva a la remisión de los síntomas del síndrome de Cushing, pero es necesario un tratamiento de sustitución permanente. En el síndrome de Carney el pronóstico depende de las manifestaciones coexistentes.
3. En caso de cáncer suprarrenal (→más adelante) el pronóstico depende de la estadificación del cáncer y de la extensión de la cirugía. En enfermos tras la adrenalectomía bilateral es necesario el tratamiento de sustitución crónico.
FIGURASArriba
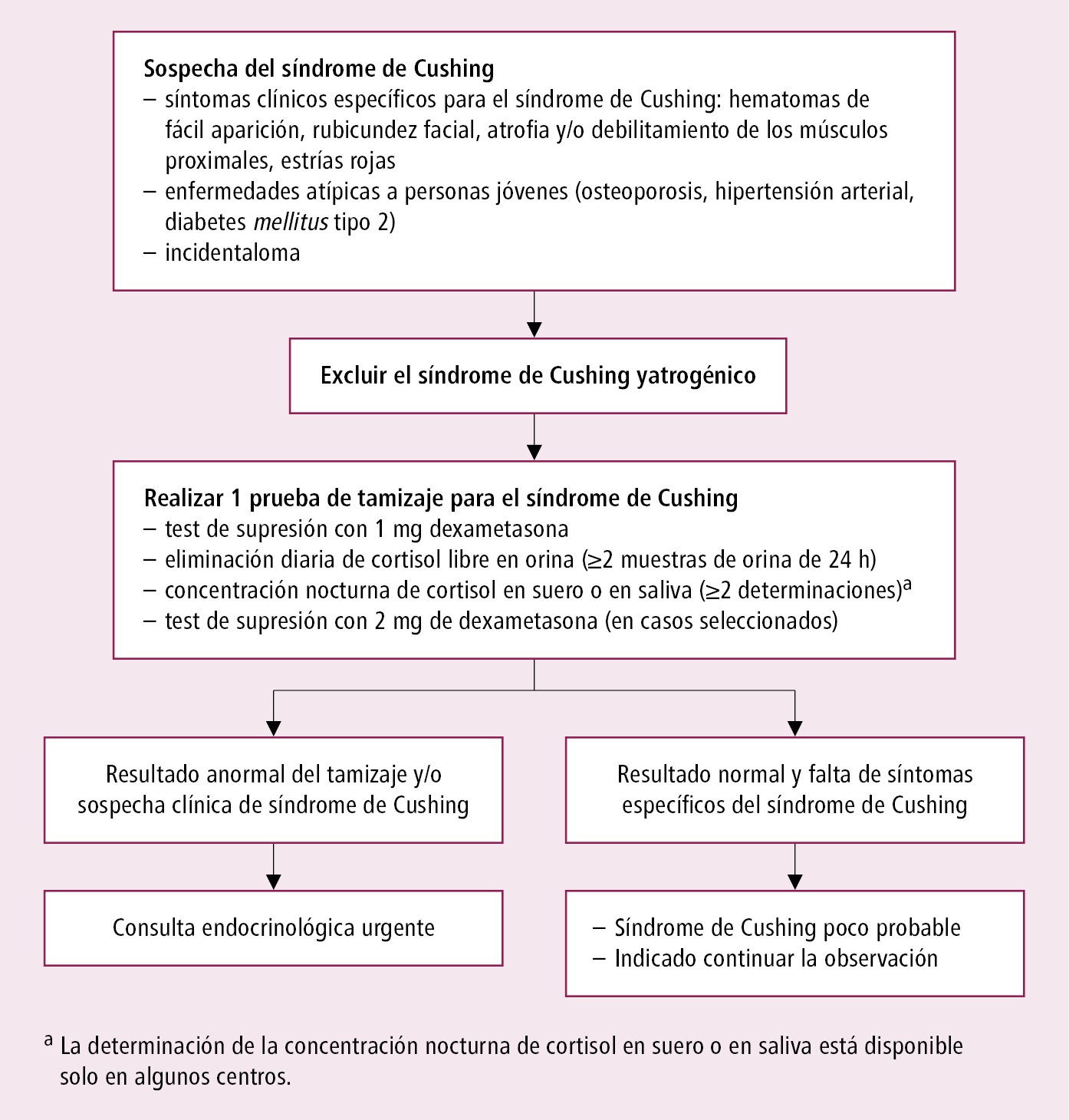
Fig. 11.2-1. Algoritmo diagnóstico en caso de sospecha del síndrome de Cushing

Fig. 11.2-2. Algoritmo diagnóstico para identificar la etiología del síndrome de Cushing
 Español
Español
 English
English
 українська
українська

