Definición y etiopatogeniaArriba
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune en la que trastornos complejos del sistema inmunitario dan lugar a un proceso inflamatorio crónico en numerosos órganos y tejidos. Etiología desconocida.
Cuadro clínico e historia naturalArriba
Las mujeres enferman 6-10 veces más frecuentemente que los hombres. Cerca de 2/3 de los casos ocurren entre los 16 y 55 años de edad. La enfermedad puede comenzar con escasos síntomas. A menudo dominan los síntomas generales o limitados a un solo órgano o sistema. Cursa con períodos de exacerbación y remisión, y en un 10-40 % de los enfermos los períodos de remisión son prolongados (>1 año). Sin embargo, en ~70 % de los enfermos, a pesar de una remisión inicial o una baja actividad de la enfermedad, se producen recidivas. La enfermedad suele ser más grave en los hombres y adolecentes En personas mayores el curso es más benigno.
PERSPECTIVA LATINOAMERICANA
En América Latina, el curso de LES suele ser más grave en afrolatinoamericanos y mestizos.
1. Síntomas generales: debilidad y fatigabilidad fácil, febrícula o fiebre, pérdida de peso.
2. Manifestaciones mucocutáneas
1) Lupus eritematoso cutáneo agudo en un 60-80 % de los casos. Forma limitada: eritema malar (→fig. 17.3-1); además de las mejillas y el puente de la nariz los cambios pueden localizarse en la frente, alrededor de los ojos, cuello y escote, y se agravan con la luz solar. La fotosensibilidad suele manifestarse en las primeras 24 h de la exposición. Los cambios persisten durante largo tiempo y pueden manifestarse como lesiones generalizadas (en otras áreas expuestas del cuerpo) de carácter eritematoso, maculopapular o folicular, en forma de ampollas, o asemejarse a la necrólisis epidérmica tóxica. En la etapa activa de la enfermedad a menudo se producen úlceras en la mucosa oral o nasal.
2) Lupus eritematoso cutáneo subagudo (LECS) en ~20 % de los casos. Los cambios aumentan o se producen bajo la influencia de la luz solar adoptando una distribución concéntrica, a menudo en forma de erupciones sobreelevadas con centro claro o erupciones foliculares descamativas (similares a las psoriásicas), generalmente en el cuello, los hombros, el tórax. No dejan cicatrices, pero pueden dejar trastornos de la pigmentación y telangiectasias.
3) Lupus eritematoso cutáneo crónico (lupus discoideo): se desarrolla en ~25 % de los enfermos; mayoritariamente limitado a la piel. Los cambios discoideos se presentan con mayor frecuencia en el cuero cabelludo, rostro, cuello y orejas, dejando cicatrices deformantes.
4) Otras lesiones cutáneas no específicas: alopecia y cabello ralo, liquen mixedematoso, anetodermia, exantema pustuloso.
5) Cambios de origen vascular: suelen deberse a vasculitis y/o microtrombosis; fenómeno de Raynaud (en un 15-40 % de los enfermos), livedo reticular, úlceras, necrosis, urticaria, eritema palmar, telangiectasias periungueales, eritromelalgia, "equimosis" que se asemejan a una astilla clavada debajo de las uñas (son microtrombosis), nódulos de Osler y lesiones de Janeway.
3. Cambios en el aparato locomotor: artralgias y/o mialgias (migratorias, de carácter variable, sobre todo en las articulaciones de manos y rodillas, en >2/3 de los enfermos); artritis y/o miositis (raramente), tendinitis y tenosinovitis. Generalmente no se producen daños de las estructuras articulares ni deformidades (la artropatía de Jaccoud es una forma rara de artropatía que cursa con deformidad articular, pero a diferencia de la AR no cursa con erosiones); osteoporosis, osteonecrosis séptica.
4. Manifestaciones renales (nefritis lúpica): las alteraciones en el análisis de orina (nefritis lúpica clínica) se observan en ~50 % de los enfermos, principalmente como resultado del depósito de inmunocomplejos en los riñones. La nefritis lúpica puede cursar como una glomerulonefritis crónica, como una glomerulonefritis rápidamente progresiva que puede condicionar un síndrome nefrítico, o bien como una nefritis intersticial con tubulopatía que puede cursar como acidosis tubular renal distal, a menudo con hiperpotasemia.
5. Cambios en el sistema respiratorio: pleuritis seca o exudativa (en ~50 % de los enfermos); raramente neumonía intersticial linfoidea aguda, que presenta una mortalidad de hasta el 50 %, y los enfermos que sobreviven desarrollan insuficiencia respiratoria grave de tipo restrictivo; hemorragia alveolar difusa; fibrosis pulmonar intersticial crónica; hipertensión pulmonar. Se deben tener en cuenta las complicaciones respiratorias del tratamiento inmunosupresor como la neumonía infecciosa y los cambios intersticiales inducidos por ciclofosfamida y metotrexato (MTX).
6. Cambios en el sistema circulatorio: pericarditis exudativa (en 50 % de los enfermos, recurrente y raramente crónica), lesiones valvulares que cursan con disfunción moderada y endocarditis no infecciosa (endocarditis de Libman-Sacks), miocarditis (poco frecuente y normalmente asintomática, generalmente detectada como una contractilidad miocárdica globalmente deprimida en ecocardiografías realizadas por taquicardia inexplicable o cambios inespecíficos del segmento ST y onda T; pueden producirse trastornos de la conducción); hipertensión arterial (como resultado de la afectación renal o como complicación de la corticoterapia), aumento del riesgo de desarrollo prematuro de ateroesclerosis y de enfermedad coronaria.
7. Cambios en el sistema nervioso (lupus neuropsiquiátrico, NPLES) en un 30-40 % de los enfermos.
1) Frecuentes (5-15 %): accidentes cerebrovasculares, tales como accidente isquémico transitorio o ACV isquémico (>80 %), ACV hemorrágico, cambios multifocales, trombosis de senos venosos, convulsiones.
2) Raras (1-5 %): alteraciones cognitivas graves, depresión, alteraciones agudas de la conciencia y afectación del sistema nervioso periférico (poli- y mononeuropatías, miastenia grave, síndrome de Guillain-Barré, plexopatías).
3) Muy raras (<1 %): síntomas psicóticos, mielopatías, corea, neuropatías de nervios craneales, incluida la neuritis inflamatoria y la neuropatía isquémica del nervio óptico, meningitis aséptica. Los síntomas pueden relacionarse con infecciones secundarias, trastornos metabólicos asociados al LES, coexistencia de síndrome antifosfolipídico, efectos secundarios de los medicamentos utilizados, sobre todo de los glucocorticoides.
8. Manifestaciones hematológicas: linfadenopatías en ~50 % de los enfermos (en general localizadas en el cuello, axilas y región inguinal). Se trata de adenopatías de hasta varios centímetros, generalmente blandas, no dolorosas ni adheridas a planos profundos; esplenomegalia, rara vez púrpura trombótica trombocitopénica secundaria.
9. Cambios en el aparato digestivo: disfagia (infrecuente, suele deberse a trastornos de la motilidad esofágica), hepatomegalia (en ~1/2 de los enfermos, puede ser en forma de hepatitis autoinmune), puede producirse peritonitis aséptica, vasculitis/trombosis de los vasos mesentéricos y pancreáticos.
DiagnósticoArriba
Exploraciones complementarias
1. Pruebas de laboratorio
1) Análisis de sangre: VHS elevada, proteína C-reactiva frecuentemente normal o solo ligeramente elevada. Se produce un aumento moderado de la proteína C-reactiva en los casos de exacerbación con serositis, en los demás casos con la proteína C-reactiva elevada hay que buscar infecciones. Anemia normocrómica (anemia de trastorno crónico), menos frecuente anemia hemolítica con test de Coombs directo positivo; leucopenia (en un 15-20 % de los enfermos) y linfopenia <1500/ml (la leucocitosis suele deberse a la infección o uso de glucocorticoides a dosis elevadas); trombocitopenia (manifestación de trastornos del sistema inmunitario en el curso de LES y/o del síndrome poliglandular autoinmune secundario, a menudo es difícil diferenciar entre los dos); pancitopenia en el curso del síndrome de activación de macrófagos (raramente), secundaria a infecciones, neoplasias o LES activo; trastornos de la coagulación debidos a la presencia de anticuerpos contra factores de coagulación o anticuerpos antifosfolipídicos; aumento de la creatinina y urea en el suero (en nefritis lúpica); hipoalbuminemia e hipergammaglobulinemia; aumento de las transaminasas en el suero.
2) Análisis de orina: proteinuria (en un 95 % de los enfermos con nefritis lúpica; puede ser de carácter nefrótico); en el sedimento urinario se aprecian hematíes dismórficos, leucocitos, cilindros eritrocitarios, leucocitarios y granulosos (el denominado sedimento activo), hematuria (raramente).
3) Pruebas inmunológicas: anticuerpos AAN y aFL (especificidad de los anticuerpos anti-dsDNA y anti-Sm para LES 95-97 %). Algunos se relacionan con una mayor incidencia de ciertas afectaciones orgánicas, p. ej. anti-dsDNA — nefritis lúpica; anti-RNP — miositis; anti-Sm — afectación del SNC y nefritis lúpica; anti-Ro (SS-A) — linfopenia, linfadenopatía, LECS, síndrome de Sjögren. Otros anticuerpos como los antinucleosomas, ribosomas (anticuerpos anti-rib P), anticuerpos anti-Ku o anti-PCNA; reacciones cruzadas a las pruebas sifilíticas (en 1/3 de los enfermos, sugieren la presencia de aFL); descenso de los factores C3 o C4 del complemento (a menudo en enfermos con nefritis lúpica). En el lupus eritematoso inducido por fármacos anticuerpos antihistonas (>75 %) y con menor frecuencia anti-ssDNA (habitualmente asintomático).
2. Pruebas histológicas: al microscopio de inmunofluorescencia las muestras de piel con lesiones eritematosas e incluso sin lesiones visibles ponen de manifiesto depósitos de inmunoglobulinas y componentes del sistema del complemento en forma de bandas a nivel de la interfase epidérmico-dérmica. Estos depósitos pueden apreciarse en otras enfermedades cutáneas y hasta en el 20 % de las personas sanas. La biopsia renal está indicada en la mayoría de los enfermos con manifestaciones de nefropatía lúpica, especialmente con proteinuria >0,5 g/d persistente. Permite especificar el tipo de glomerulopatía, la actividad y el tiempo de evolución de la enfermedad en el riñón, lo que es importante a la hora de elegir el tratamiento y establecer el pronóstico.
3. Otras: pruebas de imagen para detectar cambios en los órganos (dependiendo del cuadro clínico), examen del líquido cefalorraquídeo, EEG, estudios de conducción nerviosa y muscular, pruebas neuropsicológicas en enfermos seleccionados con NPLES.
Criterios diagnósticos
El diagnóstico se basa en la presencia de síntomas clínicos típicos, y en los resultados de las exploraciones complementarias. Es necesario detectar AAN en título >1:80. Criterios de clasificación de ACR y EULAR (2018) →tabla 17.3-1.
Diagnóstico diferencial
Enfermedad mixta e indiferenciada del tejido conectivo, síndrome de Sjögren, AR incipiente, vasculitis sistémica, SAF, lupus inducido por fármacos (causas →tabla 17.3-2), fibromialgia con presencia de AAN, neoplasias hematológicas (especialmente linfomas), púrpura trombocitopénica idiopática, anemia hemolítica autoinmune, infecciones. El eritema facial a veces debe ser diferenciado de la rosácea, dermatitis seborreica, fotodermatosis, dermatomiositis. Síntomas para diferenciar conectivopatías sistémicas →tabla 17.7-2.
TratamientoArriba
Principios generales
1. El objetivo principal es prolongar la supervivencia, evitar el daño permanente de órganos y mejorar la calidad de vida relacionada con la salud (CVRS). Esto se puede lograr controlando la actividad de la enfermedad (con el fin de conseguir la remisión o baja actividad de la enfermedad y prevenir las exacerbaciones) y minimizando las comorbilidades y la toxicidad de los fármacos. En el tratamiento deben tomarse en cuenta los factores que empeoran la CVRS, tales como la fatiga, el dolor y la depresión.
2. Se hace una distinción entre el tratamiento cuyo objetivo es inducir la remisión o —de no poder conseguirse— reducir la actividad de la enfermedad lo máximo posible (→Observación), y el tratamiento cuyo objetivo es prevenir las recaídas.
3. Fármacos: preparados, contraindicaciones y principios de observación de los efectos adversos →tabla 17.1-6.
1) Antimaláricos: hidroxicloroquina ≤5 mg/kg/d, en el tratamiento de mantenimiento por lo general 200 mg/d, eventualmente cloroquina en todos los enfermos, siempre y cuando no haya contraindicaciones. En el tratamiento de lesiones cutáneas se puede valorar el uso de mepacrina.
2) Glucocorticoides: dosificación en función de la actividad de la enfermedad, tanto en la inducción de la remisión, como en las exacerbaciones y en el tratamiento de mantenimiento.
a) forma leve (síntomas generales, artritis leve, afectación de ≤9 % de la superficie cutánea recuento de plaquetas de 50 000-100 000/µl, SLEDAI ≤6, síntomas C o ≤1 síntoma B en el BILAG) y moderada (artritis, afectación de un 9-18 % de la superficie cutánea, vasculitis cutánea que afecta a ≤18 % de la superficie cutánea, recuento de plaquetas de 20 000-50 000/µl, inflamación de la membrana serosa, SLEDAI 7-12, ≥2 síntomas B en el BILAG) → inicialmente ≤0,5 mg/kg/d (en conversión a prednisona)
b) forma grave (afectación grave de órganos importantes [riñones, SNC, médula ósea, pulmones, arterias mesentéricas], recuento de plaquetas de <20 000/µl, síndrome hemofagocítico agudo, síntomas de púrpura trombocitopénica trombótica, SLEDAI >12, ≤1 síntoma A en el BILAG) → metilprednisolona iv. 250-1000 mg/d durante 1-3 días, luego VO 0,5-0,7 mg/kg/d (en conversión a prednisona) con reducción gradual de la dosis.
En el tratamiento de mantenimiento se debe intentar utilizar glucocorticoides a la dosis efectiva más baja y, si es posible, retirarlos por completo. Tras conseguir la remisión, disminuir gradualmente la dosis de glucocorticoides en 5 mg/d cada 1-2 semanas, y después de llegar a la dosis de 20 mg/d en 2,5 mg/d cada 1-2 semanas hasta la dosis más baja necesaria para controlar los síntomas. Se debe intentar reducir la dosis de mantenimiento hasta ≤7,5 mg/d (normalmente en 4-6 meses). En caso de remisión prolongada se debe considerar interrumpir el glucocorticoide y mantener el antimalárico. Para reducir la dosis de mantenimiento o interrumpir el glucocorticoide puede estar indicado utilizar un fármaco inmunomodulador/inmunosupresor.
3) Otro fármaco inmunomodulador/inmunosupresor (metotrexato, azatioprina, micofenolato de mofetilo, eventualmente con un inhibidor de la calcineurina; la elección del medicamento depende de la localización y la gravedad de las lesiones [→más adelante], los planes reproductivos, efectos adversos y del precio): debe asociarse en casos refractarios al tratamiento con hidroxicloroquina y glucocorticoides, así como para disminuir gradualmente la dosis de glucocorticoide hasta conseguir la dosis de mantenimiento o interrumpirlo. En caso de lesiones orgánicas graves hay que utilizar estos fármacos desde el principio (junto con un antimalárico y glucocorticoide). En casos muy graves (especialmente con afectación renal, cardíaca, pulmonar o del SNC) utilizar ciclofosfamida (junto con un antimalárico y glucocorticoide) que puede sustituirse por otro inmunosupresor tras conseguir la remisión.
4) Fármacos biológicos: valorar la asociación de belimumab en enfermos con enfermedad extrarrenal muy activa, con respuesta insuficiente (imposibilidad de reducir la dosis de glucocorticoide o recidivas frecuentes) al tratamiento estándar con antimaláricos, glucocorticoides y, eventualmente, inmunosupresores. En caso de lesiones orgánicas graves resistentes al tratamiento inmunosupresor estándar, o ante la existencia de contraindicaciones para el mismo, se puede considerar utilizar rituximab.
Formas de presentación de los fármacos arriba mencionados, contraindicaciones, y principios de observación del tratamiento →tabla 17.1-6.
4. Prevención de las exacerbaciones:
1) evitar la exposición a la luz solar
2) evitar las drogas que causan lupus eritematoso inducido por fármacos
3) fármacos antimaláricos
4) abandono del tabaquismo.
5. Intervenciones adicionales:
1) prevención de la osteoporosis →Osteoporosis; tener en cuenta la posibilidad de aparición de osteonecrosis séptica y sus manifestaciones
2) combatir los factores de riesgo de enfermedad cardiovascular
3) vacunas (solo cuando la enfermedad no está activa), especialmente indicadas las vacunas antigripal y antineumocócica, otras vacunas en función de la evaluación del riesgo individual; el uso de las vacunas vivas generalmente está contraindicado cuando la dosis de glucocorticoides es >20 mg/d o se utilizan otros inmunosupresores
4) informar a mujeres en edad reproductiva tratadas con fármacos inmunosupresores sobre la necesidad de usar métodos anticonceptivos eficaces (una contraindicación de la contracepción hormonal con estrógenos es la presencia de aFL/SAF)
5) en enfermos con aFL/SAF considerar la necesidad de prescribir la profilaxis antitrombótica primaria o secundaria →Síndrome antifosfolipídico.
Tratamiento de lesiones cutáneas
1. Evitar la exposición a la luz solar: ropa de protección, protector solar con un factor de protección ≥15.
2. Tratamiento tópico: pomadas y cremas que contienen glucocorticoides (durante corto tiempo porque los derivados de flúor inducen atrofia de la piel) o un inhibidor de calcineurina (p. ej. tacrolimus al 0,1 %).
3. Fármacos antimaláricos: hidroxicloroquina 200 mg/d, eventualmente cloroquina 250 mg/d. Si esto no es eficaz, añadir mepacrina, y en caso de retinopatía cambiar por mepacrina.
4. En casos refractarios: MTX 10-25 mg 1 × semana, retinoides (p. ej. isotretinoína, inicialmente 0,5 mg/kg 2 × d, a continuación 0,25-0,5 mg/kg/d; atención: fármaco teratogénico), dapsona, MFM, ácido micofenólico, fármacos biológicos (p. ej. rituximab, belimumab), talidomida (solo como terapia de rescate ante la ineficacia de otros fármacos).
Tratamiento de trastornos hematológicos
1. Anemia inmunohemolítica y trombocitopenia inmune: por lo general responden bien al tratamiento con glucocorticoides (a dosis moderada o alta, preferiblemente iv.) combinados con un inmunosupresor (azatioprina, MFM, ciclosporina). Se puede administrar IGIV (1 g/kg/d durante 1-2 días) en fase aguda o ante una respuesta insuficiente a glucocorticoides a dosis alta, así como para evitar el riesgo de infección asociada a la terapia con glucocorticoides. En casos refractarios valorar el uso de rituximab o ciclofosfamida, y como último recurso la trombopoyetina o esplenectomía.
2. Leucopenia: generalmente no requiere tratamiento. Por lo general responde bien a glucocorticoides. En caso de neutropenia inducida por fármacos reducir la dosis o suspender el fármaco citotóxico, y en caso de agranulocitosis <500/µl administrar G-CSF. En caso de linfopenia considerar la profilaxis de la infección P. jiroveci (cotrimoxazol).
3. Púrpura trombocitopénica trombótica →Púrpura trombocitopénica trombótica.
4. Síndrome de activación de macrófagos →Situaciones especiales.
Tratamiento de artritis, artralgias y mialgias
Utilizar AINE, glucocorticoides (muy eficaces, p. ej. prednisona hasta 15 mg/d VO), hidroxicloroquina, eventualmente cloroquina (→más arriba) o MTX (10-25 mg 1 × semana con ácido fólico). En caso de dolor persistente limitado a 1 o 2 articulaciones, considerar una osteonecrosis séptica.
Tratamiento de las serositis
Usar AINE o glucocorticoides (generalmente prednisona ~15 mg/d). También son eficaces los fármacos antimaláricos, MTX, azatioprina y belimumab. En pericarditis es eficaz la colchicina.
Tratamiento de la nefritis lúpica
1. Siempre eliminar los factores que aceleran el avance de la nefropatía y actuar sobre la prevención de la enfermedad cardiovascular.
2. En enfermos con proteinuria usar IECA/ARA-II. Tratamiento del síndrome nefrótico →Síndrome nefrótico.
3. En todos los enfermos con nefropatía lúpica, incluso en remisión, se recomienda utilizar hidroxicloroquina 200-400 mg/d (máx. 5 mg/kg; se puede utilizar cloroquina 250-500 mg/d).
4. Tratamiento inmunosupresor.
El manejo depende del tipo de nefritis lúpica:
1) clase I (cambios mesangiales mínimos, no hay signos clínicos de nefropatía) → terapia inmunosupresora solo en caso de indicaciones asociadas a lesiones extrarrenales en el curso de LES
2) clase II (proliferación mesangial con acúmulos en el mesangio): sin tratamiento inmunosupresor; progresión de la nefropatía, es decir proteinuria >1 g/d o disminución de la TFG sin causa evidente → indicación de una nueva biopsia renal
3) clases III (cambios proliferativos focales en los glomérulos) y IV (cambios proliferativos difusos [≥50 % de los glomérulos]) → necesitan tratamiento inmunosupresor intensivo (glucocorticoides + ciclofosfamida o MFM), debido al mal pronóstico y progresión a una insuficiencia renal terminal
4) clase V (glomerulonefritis membranosa; el pronóstico es en general bueno) → fármacos inmunosupresores en caso de proteinuria nefrótica persistente
5) clase VI (glomeruloesclerosis avanzada) → sin terapia inmunosupresora; preparación para la terapia de sustitución renal; en la insuficiencia renal terminal el trasplante renal es el tratamiento de elección (es posible en enfermos que no han tenido evidencia de actividad de la enfermedad ≥6 meses).
Etapas de tratamiento de la glomerulopatía proliferativa (clase III y IV) y de la glomerulonefritis membranosa (clase V):
1) Inducción de la remisión de la fase aguda (3-6 meses):
a) glucocorticoides: metilprednisolona iv. 250-500 mg durante 3 días consecutivos, seguida de prednisona VO 0,6-1 mg/kg del peso ideal/d y después una reducción gradual en período de 3 meses hasta una dosis de mantenimiento ≤7,5 mg/d; y
b) ciclofosfamida 0,5 g en infusión iv. cada 2 semanas durante 3 meses (si existen factores de mal pronóstico [empeoramiento de la función renal, presencia de semilunas o necrosis fibrinoide en >25 % de los glomérulos], pueden administrarse dosis más altas: 500-1000 mg/m2 de la sc. cada mes durante 6 meses) o MFM VO 2-3 g/d (o ácido micofenólico 1440-2160 mg/d) durante 3-6 meses. En caso de progresión de la enfermedad en los primeros 3 meses de la terapia de inducción realizar un cambio al tratamiento alternativo: ciclofosfamida a MFM o viceversa.
Criterios de eficacia del tratamiento. Remisión completa: reducción de la proteinuria hasta <0,5-0,7 g/d y una disminución de la creatininemia hasta el valor inicial antes de la enfermedad. Remisión parcial: estabilización o disminución de la creatininemia (sin retorno al valor de inicio antes de la enfermedad) y reducción de la proteinuria ≥50 %. El fallo de remisión completa después de 12 meses del tratamiento generalmente es una indicación para la repetición de la biopsia renal. Un aumento de proteinuria o creatininemia durante la reducción de dosis de fármacos es una indicación para volver a aumentarlos hasta la dosis que proporcionaba el control de la enfermedad. En casos de curso especialmente grave o refractarios a la terapia de inducción estándar se debe considerar la administración de un fármaco biológico dirigido contra los linfocitos B (rituximab 2 dosis de 1 g en un intervalo de 2 semanas). En caso de contraindicaciones importantes para el uso de glucocorticoides a dosis altas o de tratamiento inmunosupresor, se pueden administrar infusiones iv. de inmunoglobulinas (en dosis total de 2 g/kg).
2) Tratamiento de mantenimiento (≥3 años): tiene como objetivo la prevención de las recurrencias o mantener la menor actividad posible de la enfermedad: azatioprina VO 2 mg/kg/d o MFM VO 2,0 g/d y glucocorticoides a dosis bajas (p. ej. prednisona VO 2,5-5 mg/d).
Tratamiento de lupus neuropsiquiátrico
1. Glucocorticoides y otros fármacos inmunosupresores (por lo general ciclofosfamida) se utilizan solo cuando la manifestación de NPLES es consecuencia de un proceso inmune (por lo general acompañado de una alta actividad sistémica del LES); siempre excluir la infección, el impacto de fármacos, los trastornos metabólicos y la neoplasia.
2. Si los síntomas de NPLES se deben a los aFL, adicionalmente utilizar fármacos antiagregantes y/o anticoagulantes →Síndrome antifosfolipídico.
3. Es necesario aplicar un tratamiento sintomático (p. ej. fármacos antiepilépticos, antidepresivos) y además tratar los factores que agravan el curso del LES (p. ej. hipertensión arterial, trastornos metabólicos, infecciones).
Tratamiento de lupus eritematoso inducido por fármacos
1. La interrupción del medicamento que ha desencadenado los síntomas suele conllevar la remisión de los síntomas en pocos días.
2. En raras ocasiones es necesario utilizar AINE y hidroxicloroquina (o cloroquina) durante un tiempo.
3. Glucocorticoides: principalmente para un control rápido de la inflamación de las membranas serosas.
4. Los enfermos con lupus inducido por hidralazina a menudo requieren tratamiento inmunosupresor.
ObservaciónArriba
1. El objetivo del tratamiento es la remisión (SLEDAI 0 ptos. en caso de tratamiento con un antimalárico, sin glucocorticoides ni un fármaco inmunosupresor) o por lo menos lograr una baja actividad de la enfermedad. Todos los enfermos deben someterse a control cada 6-12 meses, y en casos de formas moderadas o graves cada 1-3 meses.
2. Para la evaluación de actividad de LES y el diagnóstico de agudizaciones utilizar: aparición de nuevos síntomas clínicos (gravedad y tipo de lesiones de la piel, artritis, serositis, síntomas neurológicos o psicóticos), parámetros de laboratorio (hemograma, concentración de creatinina y albúmina en el suero, proteinuria, sedimento urinario, concentración de componentes C3 y C4 del complemento, títulos de anticuerpos anti-C1q y anti-dsDNA en el suero) e indicadores de la actividad general de la enfermedad (p. ej. SLEDAI).
3. Antes del embarazo, cirugía o tratamiento con estrógenos se deben determinar los aFL.
4. En función del riesgo individual, especialmente antes de una terapia inmunosupresora intensiva, realizar pruebas de detección de VHB, VHC, CMV y Mycobacterium tuberculosis.
5. Observación de los efectos adversos de los fármacos →tabla 17.1-6.
Situaciones especialesArriba
Síndrome de activación de macrófagos (SAM)
Una de las formas adquiridas de linfohistiocitosis hemofagocítica →Linfohistiocitosis hemofagocítica, que se produce en enfermedades reumáticas, más comúnmente en la forma sistémica de artritis idiopática juvenil, y en adultos con LES. Se trata de una actividad excesiva y prolongada de los macrófagos y las células T (principalmente células CD8+), que conduce a una respuesta inflamatoria no controlada. Se producen: citopenias, hiperferritinemia, fiebre habitualmente elevada, hepatomegalia, esplenomegalia o adenopatía, síntomas neurológicos.
Se diagnostica según los criterios generales de la linfohistiocitosis hemofagocítica, que sin embargo no se han verificado en la población de enfermos con LES. Requiere la diferenciación con otras formas secundarias de la linfohistiocitosis hemofagocítica, causadas p. ej. por una infección o neoplasia maligna, también con sepsis y las exacerbaciones del LES.
En el tratamiento se utilizan glucocorticoides a dosis altas (~50 % de eficacia), en caso de corticorresistencia se utiliza ciclosporina, ciclofosfamida o tacrolimus (80 % de remisión). Si el estado del enfermo empeora, se asocia etopósido junto con dexametasona y ciclosporina. Son factores de mal pronóstico la infección y proteína C-reactiva >50 mg/l.
Nefropatía asociada a síndrome antifosfolipídico en el curso del LES
La microangiopatía trombótica en el curso del LES puede manifestarse en forma de nefropatía crónica de progresión lenta que cursa sin síntomas de glomerulopatía importante (sin proteinuria o con proteinuria leve). Se presenta hipertensión arterial y pérdida progresiva de la función renal. Se observan características de microangiopatía trombótica en >20 % de los enfermos con nefropatía lúpica. Esta puede conducir a la insuficiencia renal a pesar de emplear un tratamiento eficaz y controlar la glomerulopatía lúpica. Tratamiento: hidroxicloroquina (o cloroquina) y anticoagulantes (como en el síndrome antifosfolipídico →Síndrome antifosfolipídico).
Embarazo
El LES no afecta la fertilidad, pero supone un riesgo tanto para el embarazo, como para la salud de la mujer y el recién nacido. Se recomienda diferir el embarazo hasta alcanzar la remisión de la enfermedad, incluida la nefropatía lúpica. El fracaso obstétrico y la aparición de preeclampsia se relacionan principalmente con la presencia de aFL y de nefritis lúpica. El embarazo puede aumentar la actividad del LES, pero los episodios son generalmente leves. Se debe comentar los riesgos a las mujeres que planifican el embarazo y programar las pruebas de control ≥1 vez en cada trimestre del embarazo. Antes del embarazo hay que determinar los anticuerpos anti-La, anti-Ro y aFL. En enfermos con afectación renal es necesario medir la presión arterial, determinar la concentración de creatinina sérica y el cociente proteína/creatinina en orina por lo menos cada 4-6 semanas hasta la semana 28 del embarazo, y luego cada 1-2 semanas hasta la semana 36, continuando una vez a la semana hasta el parto. No se debe interrumpir el tratamiento de mantenimiento durante el embarazo ni ≥3 meses después de su resolución. Se consideran admisibles en enfermas embarazadas los siguientes fármacos: glucocorticoides (prednisona <15 mg/d), azatioprina, cloroquina, hidroxicloroquina, ciclosporina, tacrolimus (está contraindicado el uso de ciclofosfamida, MTX y MFM) y AAS a dosis bajas. Este último debe considerarse en todas las embarazadas con nefropatía lúpica para disminuir el riesgo de desarrollar preeclampsia. Los AINE pueden utilizarse en el 1.er y 2.º trimestre (no administrar inhibidores selectivos de COX-2). Debido a los escasos datos de seguridad, los FARME biológicos (incluido el rituximab, el tocilizumab y el belimumab), deben retirarse antes del embarazo planificado, a menos que ninguno de los fármacos considerados seguros permita un control adecuado de los síntomas de la enfermedad. En el período perinatal puede ser necesario aumentar la dosis de glucocorticoides. La presencia de anticuerpos anti-Ro (anti-SS-A) y anti-La (anti-SS-B) en la embarazada puede causar lupus eritematoso neonatal (3 % de todos los embarazos) y ser causa de bloqueo cardíaco congénito en el feto. Es necesaria una monitorización estrecha durante el embarazo.
Durante la lactancia, si no hay contraindicaciones por parte del niño, puede continuarse el tratamiento con la hidroxicloroquina, cloroquina, azatioprina, ciclosporina, prednisona (a dosis de >50 mg puede amamantarse pasadas >4 h desde la toma del fármaco), inmunoglobulinas, tacrolimus, AINE no selectivos y celecoxib. No administrar MTX, MFM, ciclofosfamida ni inhibidores de COX-2 diferentes al celecoxib.
Intervenciones quirúrgicas
Antes de la cirugía valorar la actividad de la enfermedad, ya que la intervención quirúrgica puede exacerbar su curso. Se recomienda alcanzar primero la remisión, si al aplazar la intervención no se pone en peligro la vida del enfermo. La mejoría puede obtenerse rápidamente, mediante el uso de glucocorticoides a dosis altas.
PronósticoArriba
Causas más comunes de muerte en las primeras etapas de la enfermedad: infección y lesiones graves en órganos (afectación del SNC, del sistema cardiovascular, neumonitis lúpica aguda, nefropatía grave); posteriormente complicaciones del tratamiento (infección) y consecuencias de la ateroesclerosis acelerada, trombosis. La progresión del daño orgánico depende en gran medida de la aparición de hipertensión arterial y del uso de glucocorticoides; la hidroxicloroquina tiene una influencia positiva. Con un diagnóstico y tratamiento adecuados ~80 % de los enfermos sobrevive 10 años, y un 65 % 20 años. En más de la mitad de los casos se producen lesiones orgánicas permanentes. El pronóstico es peor en enfermos con nefritis lúpica; a pesar del tratamiento un 10-20 % de los enfermos desarrolla insuficiencia renal terminal. Las recurrencias de LES en riñón trasplantado son extremadamente raras (2 %).
TABLAS Y FIGURASArriba
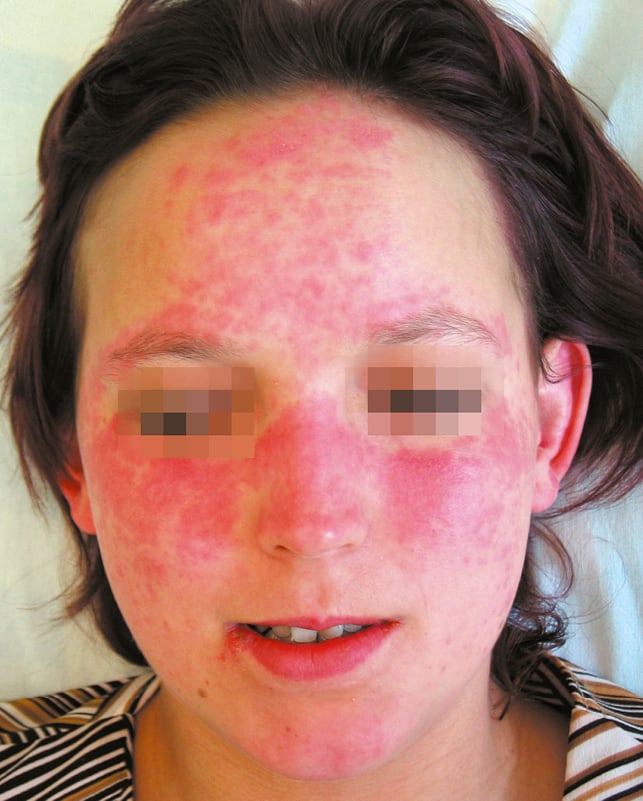
Fig. 17.3-1. Lupus eritematoso sistémico, típico eritema malar
Tabla 17.3-1. Criterios de clasificación del lupus eritematoso sistémico según ACR y EULAR (2018)
|
Dominios clínicos |
Puntuación |
|
General
Fiebre (>38,3 °C) |
2 |
|
Piel
– alopecia no cicatrizal
– úlceras orales
– lupus cutáneo subagudo o discoide
– lupus cutáneo agudo |
2
2
4
6 |
|
Artritis
Sinovitis o dolor a la palpación de ≥2 articulaciones y rigidez matutina ≥30 min |
6 |
|
Neurológico
– delirio
– psicosis
– convulsiones |
2
3
5 |
|
Serositis
– derrame pleural o pericárdico
– pericarditis aguda |
5
6 |
|
Hematológico
– leucopenia (<4000/μl)
– trombocitopenia (<100 000/µl)
– hemólisis autoinmunitaria |
3
4
4 |
|
Renal
– proteinuria >0,5 g/24 h
– biopsia renal: nefritis lúpica de clase II o V
– biopsia renal: nefritis lúpica de clase III o IV |
4
8
10 |
|
Dominios inmunológicos |
|
Anticuerpos antifosfolipídicos (aFL)
Anticardiolipina (IgA, IgG o IgM) a títulos moderados o altos (>40 U APL/GPL/MPL o percentil >99), anti-β2 glicoproteína I (β2GPI en clase IgA, IgG o IgM) o anticoagulante lúpico |
2 |
|
Sistema del complemento
– baja concentración de C3 o C4
– baja concentración de C3 y C4 |
3
4 |
|
Anticuerpos altamente específicos
– anti-ADN de doble cadena
– anti-Sm |
6
6 |
|
Nota:
1) Para cumplir los criterios es necesario conseguir >10 ptos. (y cumplir >1 criterio clínico) y el título de AAN >1:80 en células epiteliales humanas o resultado positivo de una prueba equivalente.
2) En cada dominio se toma en cuenta solo el criterio de mayor puntuación.
3) No se toman en consideración los criterios cuya presencia puede explicarse mejor por otra enfermedad.
4) Los criterios no tienen que aparecer simultáneamente. |
Tabla 17.3-2. Medicamentos que causan lupus inducido por fármacosa
|
Función |
Ejemplos |
|
Antiarrítmicos |
Procainamida, quinidina, acebutolol, labetalol, disopiramida |
|
Antihipertensivos |
Hidralazina, dihidralazina, metildopa, captopril |
|
Inmunomoduladores |
Sulfasalazina, inhibidores de TNF (infliximab, adalimumab, etanercept), interferón α, inhibidores de puntos de control, leflunomida |
|
Quimioterapéuticos antineoplásicos |
Mitotano, gemcitabina, capecitabina, hidroxiurea |
|
Antimicrobianos |
Minociclina, isoniazida, terbinafina |
|
Antiepilépticos |
Carbamazepina, fenitoína |
|
Otros |
Penicilamina, clorpromazina, propiltiouracilo, IBP |
|
a Los inhibidores de TNF y los quimioterapéuticos antineoplásicos, debido a su mayor utilización, son los fármacos más frecuentemente implicados, pero los medicamentos que se asocian a un mayor riesgo de lupus inducido por fármacos son la procainamida (en un 15-20 % de los enfermos que lo usan durante 1 año) y la hidralazina (5-10 %). En el caso de algunos fármacos (procainamida, hidralazina), enferman con mayor frecuencia las personas con menor actividad congénita de la N-acetiltransferasa. |
Tabla 17.7-2. Signos diferenciales de las enfermedades del tejido conectivo
|
Signo clínico |
LES |
AR |
ES |
PM |
EMTC |
|
Pleuritis o pericarditis |
++++ |
+ |
+ |
– |
+++ |
|
Artritis destructiva |
± |
++++ |
+ |
± |
+ |
|
Fenómeno de Raynaud |
++ |
– |
++++ |
+ |
++++ |
|
Miositis |
+ |
+ |
+ |
++++ |
+++ |
|
Acroesclerosis |
± |
– |
++++ |
– |
++ |
|
Endurecimiento de la piel |
– |
– |
+++ |
– |
– |
|
Fibrosis pulmonar intersticial |
+ |
+ |
+++ |
++ |
+++ |
|
Hipertensión pulmonar |
+ |
± |
++ |
+ |
+++ |
|
Eritema ("en alas de mariposa") en la cara |
++++ |
– |
– |
– |
++ |
|
Úlceras en la boca |
+++ |
– |
– |
– |
++ |
|
Convulsiones o psicosis |
+++ |
– |
– |
– |
– |
|
Neuropatía del trigémino |
+ |
– |
++ |
– |
+++ |
|
Polineuropatía periférica |
++ |
++ |
± |
– |
++ |
|
Mielitis transversa |
+++ |
+ |
– |
– |
++ |
|
Meningitis aséptica |
+++ |
+ |
– |
– |
+++ |
|
Glomerulonefritis |
|
|
proliferativa |
++++ |
– |
– |
– |
+ |
|
|
membranosa |
+++ |
– |
– |
– |
++ |
|
Hipertensión renovascular |
+ |
– |
++++ |
– |
+++ |
|
Vasculitis sistémica |
++ |
++ |
+ |
+ |
+ |
|
Vasculopatía no inflamatoria |
– |
– |
++++ |
– |
+++ |
|
Trastornos de la motilidad esofágica |
± |
± |
++++ |
++ |
+++ |
|
Anti-RNP |
++ |
– |
+ |
+ |
++++ |
|
Anti-Sm |
+++ |
– |
– |
– |
– |
|
Anti-dsDNA |
++++ |
– |
– |
– |
– |
|
Anti-Scl 70, ACA |
– |
– |
+++ |
– |
– |
|
↓ de niveles de componentes del complemento |
+++ |
– |
– |
– |
+ |
|
FR |
++ |
+++ |
+ |
+ |
++ |
|
La cantidad de signos “+” indica la frecuencia de aparición. |
Tabla 17.1-6. Fármacos modificadores de la enfermedad (FARME) utilizados en la AR
|
Fármaco |
Dosificación |
Contraindicaciones |
Efectos adversos |
Observación |
|
Convencionales sintéticos (csFARME) |
|
Cloroquina
|
VO 250 mg 1 × d |
Enfermedades de la retina, alteraciones visuales, insuficiencia renal, porfiria, psoriasis, deficiencia de glucosa-6-fosfato deshidrogenasa, hepatitis B o C no tratada con insuficiencia hepática en clase C de Child-Pugh |
Daño retiniano (de la mácula lútea), especialmente “en ojo de buey”; exantema; dolor abdominal, diarrea, pérdida de apetito, náuseas; otros, muy raros, como miopatía, visión borrosa, trastornos de acomodación, cambios en la pigmentación de la piel y de las membranas mucosas, neuropatía periférica, miocardiopatía |
Examen oftalmológico con campimetría y tomografía de coherencia óptica de dominio espectral (SD-OCT) al inicio y, si no hay factores de riesgo de retinopatíaa, después de 5 años y luego una vez al año. Por ficha técnica, se debe realizar el examen oftalmológico (oftalmoscopia y campimetría) antes del tratamiento, posteriormente cada 3-4 meses y ante la aparición de cualquier alteración visual. |
|
Hidroxicloroquina |
VO 200 mg 1-2 × d
(≤5,0 mg/kg/d) |
Véase más arriba; aceptable en casos seleccionados con hepatitis B activa |
Véase más arriba; el riesgo de lesión ocular es muy bajo en los primeros 5-7 años del tratamiento (es mayor en casos de edad avanzada, enfermedad renal, antecedentes de retinopatía) |
Examen oftalmológico con campimetría y tomografía de coherencia óptica de dominio espectral (SD-OCT) al inicio y, si no hay factores de riesgo de retinopatíaa, después de 5 años y posteriormente una vez al año. No requiere pruebas rutinarias de laboratorio |
|
Ciclosporina
|
VO 2,5 mg/kg/d dividida en 2 dosis cada 12 h, y luego incrementar en 0,5 mg/ kg/d cada 2-4 semanas hasta obtener mejoría clínica o hasta una dosis total de 5 mg/kg/d |
Insuficiencia renal; hipertensión arterial; infecciones crónicas |
Insuficiencia renal; hipertensión arterial; anemia; vellosidad excesiva, hirsutismo en mujeres; trastornos de la sensibilidad; hiperplasia gingival; inmunodeficiencia con riesgo aumentado de infecciones
Nota: muchos medicamentos interfieren con la ciclosporina, lo que aumenta el riesgo de efectos adversos |
ECG al inicio; presión arterial y glucemia durante cada visita; creatinina en el suero cada 2 semanas hasta establecer la dosis, después cada mes; la disfunción renal causada por ciclosporina es reversible en gran medida, pero no completamente; hemograma, ALT y AST, albúmina (como más adelante): considerar la monitorización de la concentración sérica del fármaco |
|
Leflunomida
|
VO 20 mg 1 × d |
Infecciónb; leucopenia <3000/µl; trombocitopenia <50 000/µl; mielodisplasia; neoplasia linfoproliferativa tratada en los últimos ≤5 años; daño hepáticoc,d,e; embarazo y lactancia; disfunción renal grave o moderada |
Diarrea, dolor abdominal, náuseas; exantema; alopecia; daño hepático; daño renal; aumento de presión arterial; efecto teratogénico (se necesita un método anticonceptivo eficaz); en el caso de complicaciones posteriores a la interrupción de la administración del fármaco puede acelerarse su eliminación mediante la colestiramina (8 g 3 × d durante 11 días) o carbón activado (50 g 4 × d durante 11 días); en mujeres que desean quedarse embarazadas y en hombres que planean paternidad deben medirse repetidamente las concentraciones del metabolito de leflunomida de eliminación acelerada |
Hemograma, concentración de creatinina/TFGe, ALT y AST, albúmina: cada 2 semanas hasta establecer una dosis fija durante 6 semanas, a continuación cada mes durante 3 meses, luego por lo menos cada 12 semanas; con mayor frecuencia en enfermos con mayor riesgo de toxicidad; en el caso de un aumento sostenido de ALT/AST >3 × LSN hay que suspender el fármaco y considerar la biopsia hepática para evaluar daños; presión arterial y peso corporal durante cada visita |
|
Metotrexato
|
VO, IM o Vsc 10-15 mg 1 × semana, la dosis se aumenta gradualmente hasta un máx. de 25-30 mg; administración concomitantef de ácido fólico (≥5 mg/semana) o de ácido folínico con el fin de evitar los efectos adversos (citopenia, úlceras orales y náuseas) |
Véase más arriba + neumonía intersticial/fibrosis pulmonar g; aclaramiento de creatinina <30 ml/min |
Aumento de la actividad de las enzimas hepáticas en el suero, fibrosis y cirrosis hepática (muy raramente); factores de riesgo: falta de suplementación del ácido fólico, esteatohepatitis no alcohólica, sexo masculino, hiperlipidemia no tratada, creatininemia aumentada, consumo de alcohol, obesidad, diabetes, hepatitis B y C; citopenia debida a la supresión de la médula ósea y a sus complicaciones (dependiendo de la dosis); úlceras orales, incidencia de un 30 %; náuseas a las 24-48 h de la ingesta; cambios intersticiales pulmonares, frecuencia de un 2-6 %, independientemente del tiempo de tratamiento y de la dosis de metotrexato; efecto teratogénico: se necesita un método anticonceptivo eficaz; el metotrexato debe retirarse (hombre y mujer) tres meses antes de un intento de concebir. Efectos más leves (por falta de ácido fólico): mucositis, alopecia, trastornos del tracto digestivo |
Véase más arriba + prueba de la función pulmonar y radiografía de tórax antes del tratamiento (vigente desde el año anterior) y durante el tratamiento, si se desarrolla tos o disnea |
|
Sulfasalazina |
VO 1 g 2 × d (óptimamente 3-4 g/d, la dosis debe aumentarse gradualmente); simultáneamente la administración de ácido fólico (5 mg/semana) o de ácido folínico |
Hipersensibilidad a sulfonamidas y a salicilatos; casos de ileostomía; daño hepáticoc,d,h; insuficiencia renal; porfiria; deficiencia de glucosa-6-fosfato deshidrogenasa |
La mayoría de los efectos adversos se produce en los primeros meses de tratamiento: se pueden evitar al comenzar con una dosis baja e incrementar gradualmente; pérdida de apetito, dispepsia, náuseas, vómitos, dolor abdominal (frecuencia de un 30 %); cefaleas y mareos; fiebre; reacciones alérgicas de la piel (urticaria, hipersensibilidad a la luz solar) y de las articulaciones; anemia hemolítica (en enfermos con deficiencia de glucosa-6-fosfato deshidrogenasa en eritrocitos), anemia aplásica esporádica; granulocitopenia (1-3 %), puede ocurrir en cualquier momento del tratamiento (por lo general en los primeros 3 meses); un aumento de actividad de ALT/AST en el suero; cambios intersticiales pulmonares raramente |
Hemograma, ALT/AST, creatinina/eTFG séricas: véase más arriba; no se requieren nuevos controles pasados 12 meses, si el estado clínico es estable |
|
Puntuales sintéticos (psFARME) |
|
Tofacitinib
Baricitinib
Upadacitinib
Filgotinib |
5 mg 2 × d (5 mg 1 × d en enfermos con insuficiencia hepática en clase B de Child-Pugh o con aclaramiento de creatinina <30 ml/min)
2-4 mg 1 × d (2 mg/d en enfermos ≥75 años, con aclaramiento de creatinina 30-60 ml/min e infecciones crónicas o recurrentes, tratados con probenecid)
15 mg 1 × d
200 mg 1 × d (100 mg 1 × d en enfermos ≥75 años o con aclaramiento de creatinina 15-60 ml/min) |
– infecciones activas o crónicas graves (también locales)
– recuento de linfocitos <500/µli
– recuento de neutrófilos <1000/µl
– concentración de hemoglobina <8 g/dlj
– insuficiencia hepática grave (clase C de Child-Pugh)
– neoplasia activa
– episodios tromboembólicos recurrentes (si no hay anticoagulación)
– embarazo y lactancia (en caso de planificar procreación se recomienda un intervalo de 4 semanas desde la última dosis)
– en enfermos >65 años utilizar tofacitinib solo si no hay otra alternativa
|
– infecciones, también oportunistas
– reactivación de infecciones virales (especialmente de varicela y herpes zóster)
– eventos tromboembólicos en enfermos con factores de riesgo de trombosis
– linfopenia, neutropenia, anemia
– hiperlipidemia |
Antes del tratamiento: como en fármacos biológicos; durante el tratamiento, entre otros:
– hemograma de sangre periférica con fórmula leucocitaria, AST/ALT y creatinina con TFGe (al inicio, después de 4 y 12 semanas de tratamiento y luego cada 3 meses)
– lipidograma al inicio y después de 3 meses de tratamiento
– evaluación para detectar cáncer de piel no melanoma antes de iniciar la terapia y cada año durante el tratamiento |
|
Biológicos (bFARME) |
|
Abatacept
|
infusión iv. 30 min; <60 kg — 500 mg, 60-100 kg — 750 mg, >100 kg — 1 g; las dosis posteriores a las 2 y 4 semanas de la primera infusión, y luego cada 4 semanas |
Infecciones b; hepatitis viralesd,h, embarazo y lactancia |
Infecciones graves (incluidas oportunistas); probablemente leucoencefalopatía multifocal progresiva (muy raramente) |
Antes del tratamiento: radiografía de tórax y test de tuberculina/test IGRA, hemograma, ALT/AST, creatinina sérica, pruebas de hepatitis; se recomienda la vacunación contra: neumococo (periódicamente), gripe (anualmente), hepatitis B (si existen factores de riesgo); vacunas vivas contraindicadas; durante el tratamiento vigilar los síntomas de infección; en mujeres indicación de mamografía antes del tratamiento |
|
Adalimumab
|
VSc 20-40 mg cada 1 o 2 semanas |
Infecciones crónicas, véase más arriba + insuficiencia cardíaca (NYHA III o IV y FEVI ≤50 %); esclerosis múltiple u otra enfermedad desmielinizante; neoplasia linfoproliferativa tratada en los últimos ≤5 años k |
Infecciones graves (incluyendo oportunistas); formación de autoanticuerpos, incluyendo: ANA, Anti-dsDNA, anticardiolipinas y antiquimeras; raras veces se desarrolla lupus inducido por fármacos: en este caso suspender el tratamiento; citopenias (principalmente leucopenia); síndromes desmielinizantes, neuritis óptica (muy raras veces): los síntomas desaparecen después de la interrupción del fármaco; reactivación de la infección por VHB; aumento de la actividad de ALT/AST en el suero |
|
Etanercept
|
VSc 25 mg 2 × semana o 50 mg 1 × semana |
|
Infliximab
|
iv. 3-10 mg/kg inicialmente en las semanas 0, 2 y 6, después cada 8 semanas o 3-5 mg/kg cada 4 semanas |
|
Golimumab |
VSc 50 mg 1 × mes |
Véase más arriba |
Véase más arriba |
Véase más arriba |
|
Certolizumab
|
VSc 200 mg 2 × d en las semanas 0, 2 y 4, a continuación una dosis de mantenimiento de 200 mg cada 2 semanas |
Véase más arriba |
Véase más arriba |
Véase más arriba |
|
Rituximab
|
iv. 1 g 2 veces en un intervalo de 14 días; se puede repetir después de 6 meses |
Infecciones b; hepatitis viralesd,h, embarazo y lactancia |
Reacciones alérgicas; infecciones; probablemente leucoencefalopatía multifocal progresiva (muy raras veces); reactivación de la infección por VHB |
Véase más arriba + concentración de inmunoglobulinas séricas |
|
Tocilizumab
|
iv. 8 mg/kg cada 4 semanas |
Infección b; hepatitis viralesd,l; ALT/AST >5 × LSN; neutropenia <500/µl, y trombocitopenia <50 000/µl; embarazo y lactancia |
Infecciones, neutropenia y trombocitopenia, aumento de la actividad sérica de ALT/AST (particularmente con el uso concomitante de fármacos potencialmente hepatotóxicos, p. ej. FARME), trastornos lipídicos, perforación intestinal en enfermos con diverticulitis (poco común) |
Véase más arriba (debido a la supresión de la respuesta de fase aguda, debe vigilarse sobre todo el desarrollo de infección) + ALT/AST cada 4-8 semanas durante los primeros 6 meses de tratamiento, y luego cada tres meses; hemograma después de 4-8 semanas de tratamiento, y luego según las indicaciones |
|
a Dosis de cloroquina >2,3 mg/kg/d, de hidroxicloroquina >5 mg/kg/d, disminución de la TFGe, uso de tamoxifeno, maculopatía.
b Infecciones que requieran hospitalización o administración parenteral de antibióticos, tuberculosis (activa o latente, si el enfermo no recibe tratamiento profiláctico contra la tuberculosis), infección activa por varicela zóster y herpes zóster, infección fúngica activa grave (en el caso de agentes biológicos también presumiblemente infección del tracto respiratorio superior viral con fiebre y úlceras de la piel infectadas, no cicatrizadas).
c Actividad de ALT y/o AST >2 × LSN.
d Hepatitis B o C aguda.
e Hepatitis B o C aguda o crónica (independientemente del tratamiento y del grado de insuficiencia hepática).
f No administrar el mismo día que el metotrexato.
g El antecedente de enfermedad pulmonar no es contraindicación absoluta para usar cualquier FARME.
h Hepatitis B crónica (salvo hepatitis tratada en la insuficiencia hepática en clase A de Child-Pugh: la sulfasalazina puede administrarse en insuficiencia hepática clase A o B) o hepatitis C crónica en clase B o C (el etanercept está indicado como potencialmente seguro en enfermos con hepatitis C crónica).
i Para tofacitinib <750/µl
j Para tofacitinib <9 g/dl
k El rituximab está indicado en enfermos con AR aptos para el tratamiento biológico, en los que (en cualquier momento) se ha tratado una neoplasia linfoproliferativa o el melanoma o que en los últimos 5 años han padecido cáncer de piel o una neoplasia sólida.
l No hay pruebas de la seguridad de uso del tocilizumab en la hepatitis viral crónica.
ALT — alanina-aminotransferasa, AR — artritis reumatoide, AST — aspartato-aminotransferasa, FEVI — fracción de eyección del ventrículo izquierdo, TFGe — tasa de la filtración glomerular estimada |
 Español
Español
 English
English
 українська
українська

