Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-875. doi: 10.1016/S2213-8587(21)00235-7. Epub 2021 Oct 20. PMID: 34687601; PMCID: PMC8743006.
Varlamov EV, Langlois F, Vila G, Fleseriu M. MANAGEMENT OF ENDOCRINE DISEASE: Cardiovascular risk assessment, thromboembolism, and infection prevention in Cushing’s syndrome: a practical approach. Eur J Endocrinol. 2021 Apr 22;184(5):R207-R224. doi: 10.1530/EJE-20-1309. PMID: 33539319.
Nieman LK, Biller BM, Findling JW, et al; Endocrine Society. Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31. doi: 10.1210/jc.2015-1818. Epub 2015 Jul 29. PMID: 26222757; PMCID: PMC4525003.
Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015 Aug 29;386(9996):913-27. doi: 10.1016/S0140-6736(14)61375-1. Epub 2015 May 21. PMID: 26004339
Definition, Etiology, PathogenesisTop
Cushing syndrome is a clinical syndrome that includes signs and symptoms resulting from longstanding exposure of tissues to excess glucocorticoids.
Cushing syndrome is classified based on etiology.
1. Exogenous Cushing syndrome is caused by administration, by any route, of glucocorticoids at doses exceeding their physiologic levels. This is the most common cause of Cushing syndrome.
2. Endogenous Cushing syndrome results from adrenal overproduction of glucocorticoids.
1) Adrenocorticotropic hormone (ACTH)–dependent Cushing syndrome (80% of cases) is caused by excess ACTH production.
a) Pituitary adenoma (70%), also known as Cushing disease.
b) Ectopic ACTH secretion (10%): Extrapituitary neuroendocrine tumors (NETs) (bronchial cancer, thymic cancer, pheochromocytoma/paraganglioma, gastrointestinal cancer, islet cell cancer, small cell lung carcinoma, medullary thyroid cancer).
c) Ectopic corticotropin-releasing hormone (CRH) secretion (0.5%).
d) Pituitary carcinoma (very rare).
2) ACTH-independent Cushing syndrome (20% of cases): Adrenal tumors: Excess cortisol inhibits CRH and ACTH secretion, causing atrophy of the adrenal cortex located outside the tumor capsule and atrophy of the contralateral adrenal gland. Adrenal tumors are more commonly unilateral than bilateral:
a) Adrenal adenoma (10%).
b) Adrenal carcinoma (10%) may lead to excess cortisol production alone or with androgen and aldosterone overproduction.
c) Bilateral macronodular adrenal hyperplasia (<1%) is characterized by large multiple nodules sized >1 cm. It is usually sporadic or, less frequently, familial (ARMC5 mutation). Pathophysiology is related to aberrant G protein–coupled receptors activated by atypical stimuli, such as gastric inhibitory peptide, catecholamines, antidiuretic hormone, thyroid-stimulating hormone, luteinizing hormone, angiotensin II, serotonin, and human chorionic gonadotropin.
d) Bilateral micronodular adrenal hyperplasia (primary pigmented nodular adrenal disease [PPNAD]) is characterized by nodule diameter <1 cm. It may be sporadic or occur as part of Carney complex (a hereditary autosomal dominant condition associated with abnormalities in various tissues such as cardiac myxoma, lentiginosis, breast ductal adenoma, and testicular tumors).
Clinical Features and Natural HistoryTop
Signs and symptoms are mainly nonspecific, but some are more distinctive and specific than others. In the list below the more specific features are marked with a hash (their presence markedly increases the probability of Cushing syndrome), and those sensitive, with an asterisk (their absence suggests that Cushing syndrome is less likely).
Symptoms:
1) General: Weight gain* and fatigue/poor exercise tolerance; polydipsia, polyuria, and polyphagia; frequent infections, especially opportunistic (eg, fungal, tuberculosis, Pneumocystis jiroveci pneumonia), often with a severe clinical course.
2) Cutaneous: Skin prone to injury with poorly healing ulcers and easy bruising in nontraumatic areas#.
3) Neurologic and musculoskeletal: Headache, cognitive impairment; bone pain or osteopenia at an early age#.
4) Psychiatric: Emotional instability*, depression*, psychotic conditions in severe disease*.
5) Cardiovascular: Symptoms of coronary artery disease, heart failure.
6) Gastrointestinal: Peptic ulcer disease (particularly in patients treated with nonsteroidal anti-inflammatory drugs [NSAIDs]).
7) Genitourinary: Decreased libido*; erectile dysfunction in men; oligomenorrhea or secondary amenorrhea in women*; acne and hirsutism in patients with androgen-secreting tumors; nephrolithiasis.
Objective findings:
1) General: Facial plethora*; facial fullness (“round face”)*; central weight gain and elevated body mass index*; hypertension*, particularly diastolic hypertension (>105 mm Hg)#.
2) Cutaneous: Wide (>1 cm) and depressed atrophic purple striae#; dorsocervical fat pad, supraclavicular fullness#; thin skin (<2 mm)*#, easy bruising#; spontaneous cutaneous hemorrhages or purpura#.
3) Neurologic and musculoskeletal: Osteopenia or fracture; proximal muscle atrophy and/or weakness#.
4) Metabolic: Hyperglycemia; hypokalemic alkalosis#.
5) Genitourinary: Hyperandrogenism (rare or mild in adrenal adenoma); in women with adrenal tumor and evident signs of androgen excess, especially with virilization (including rapidly progressive hirsutism, temporal balding, deepening voice, male body habitus, and clitoral hypertrophy), adrenal carcinoma is strongly suspected. In men, feminizing features may be observed (in very rare estrogen-secreting tumors).
Natural history:
1. Subclinical Cushing syndrome/mild excess cortisol secretion should be suspected in patients with:
1) Adrenal incidentaloma with no previous adrenal history.
2) No clear Cushingoid phenotype (absence of the most specific features of Cushing syndrome).
3) Evidence of ACTH-independent cortisol production; most often based on a 1-mg dexamethasone suppression test (DST) with other confirmatory tests of cortisol excess being normal (see Diagnosis, below). The patient may have associated diabetes, dyslipidemia, hypertension, weight gain, and/or osteoporosis. The risk of progression to overt Cushing syndrome is low in most cases.
2. Overt Cushing syndrome: Most features (particularly the specific ones) are present, as described above.
DiagnosisTop
Diagnostic algorithm: Figure 1. Always exclude prior use of glucocorticoids (exogenous Cushing syndrome) and pseudo–Cushing syndrome.
Approach to identifying the cause of endogenous Cushing syndrome: Figure 2.
Pseudo–Cushing syndrome, with high serum cortisol levels resulting from other abnormalities/systemic effects rather than from organic lesions in the pituitary-adrenal system:
1) Depression: High serum cortisol levels and impaired dexamethasone suppression with a preserved circadian rhythm of cortisol secretion and normal serum ACTH levels.
2) Pregnancy: High blood levels of transcortin (glucocorticoid-binding globulin) and, consequently, high cortisol levels. Placental CRH secretion increases in the third trimester of pregnancy. Urinary free cortisol (UFC) excretion is increased and the circadian rhythm of cortisol secretion is preserved.
3) Alcohol dependence: Few patients have clinical features of Cushing syndrome (because of altered cortisol metabolism in the liver and effects of alcohol on the central nervous system). Abstinence results in resolution of symptoms.
4) Anorexia nervosa: Elevated cortisol levels, mainly as a result of decreased renal clearance of cortisol. An increase in ACTH secretion may also occur. Impaired dexamethasone suppression caused by acquired glucocorticoid receptor resistance is present, which explains the lack of clinical features of Cushing syndrome.
5) Medications: Certain drugs (eg, carbamazepine, fenofibrate) can elevate UFC levels depending on the assay used.
6) Morbid obesity.
7) Poorly controlled diabetes.
8) High urine output >4 to 5 L/d.
9) Acute stress and/or severe systemic illness.
Usually pseudo–Cushing syndrome can be excluded clinically but if not, the following tests can be used:
1) Dexamethasone-suppressed CRH stimulation test (3-day test): Oral dexamethasone 0.5 mg every 6 hours × 8; start at noon and administer the last dose at 6 am; then at 8 am administer CRH 1 microg/kg IV and plasma cortisol 15 minutes later; cortisol level <38 nmol/L excludes true Cushing syndrome.
2) Desmopressin stimulation test (10 microg IV): The test is described below under “Tests to identify causes of or localize Cushing syndrome”. Blood ACTH and cortisol levels rise in true Cushing disease.
Biochemical evaluation is recommended in patients with:
1) Features of Cushing syndrome.
2) Incidentally found adrenal masses.
Screening in the general population is discouraged.
Before beginning biochemical evaluation for hypercortisolism, take a detailed history of drug use to exclude exogenous glucocorticoid exposure from any route (eg, nasal, inhaled, topical) that could explain the signs and symptoms. Also, ask about herbs and supplements that may contain glucocorticoids.
1. Basic biochemical tests:
1) Metabolic panel may reveal hypokalemia, hyperglycemia (impaired glucose tolerance or diabetes); high total serum cholesterol, low-density lipoprotein cholesterol (LDL-C), and triglyceride levels; and low serum high-density lipoprotein cholesterol (HDL-C) levels.
2) Complete blood count (CBC) may reveal elevated white blood cell (particularly neutrophilia) and platelet counts. Lymphocyte, eosinophil, and monocyte counts tend to be low. The hemoglobin level is variable.
2. Screening:
1) Inadequate reduction in serum cortisol levels after the 1-mg overnight dexamethasone suppression: Patients are instructed to take oral dexamethasone 1 mg at their bedtime (11 pm to midnight). Fasting serum cortisol levels are measured on the following day between 8 and 9 am, and levels <50 nmol/L (1.8 microg/dL) exclude Cushing syndrome. With any DST, keep in mind any factors that may raise or reduce dexamethasone levels, such as drug interactions (especially those involving P450) and malabsorption, which can result in inaccurate cortisol results due to affected dexamethasone bioavailability. Estrogen and active hepatitis can also elevate total cortisol levels.
2) 24-hour UFC excretion increased to 3 to 4 × upper limit of normal (ULN) suggests the diagnosis of Cushing syndrome (sensitivity and specificity are 95%-100% and 94%-98%, respectively); collection should be repeated at least twice.
3) Elevated late-night salivary cortisol levels at 11 pm to midnight (not useful in shift workers or patients with altered sleep patterns).
4) Loss of the normal circadian cortisol secretion rhythm (the late afternoon serum cortisol level, normally usually lower, is >50% of the morning level).
3. Diagnostic or confirmatory tests:
1) 24-hour UFC excretion (see above).
2) Inadequate reduction in 9 am blood serum cortisol after the last dose in the 2-day low-dose DST (2-day 2-mg test): Instruct the patient to take oral dexamethasone 0.5 mg every 6 hours for 2 days (usually at 9 am, 3 pm, 9 pm, and 3 am on each day, for a total of 8 doses). The response is considered normal when the 9 am cortisol (6 h after the last dose) is <50 nmol/L (90%-100% sensitivity).
3) Elevated late-night salivary cortisol levels (see above).
4) Loss of the normal circadian cortisol secretion rhythm (see above).
Note that different collection methods and assays available for the measurement of cortisol exist and results may vary. When performing a diagnostic workup, it is important to evaluate the assays available at each center and carefully collect samples with accurate timing for testing.
Ensure that 2 to 3 tests are performed to establish the diagnosis of Cushing syndrome before going onto localization studies.
4. Tests to identify causes of Cushing syndrome: Once hypercortisolism is documented, the next step is to determine if it is ACTH dependent (ACTH secreting) or ACTH independent (adrenal). This is done by measuring serum ACTH levels.
1) Step 1: Serum ACTH (repeat measurements). Serum ACTH levels <2.2 pmol/L indicate ACTH-independent Cushing syndrome (adrenal source), whereas serum ACTH levels >4.4 pmol/L indicate ACTH-dependent Cushing syndrome. In patients with serum ACTH levels 2.2 to 4.4 pmol/L, the CRH or 1-desamino-8-D-arginine-vasopressin (DDAVP, desmopressin) stimulation test (see below) may be useful to distinguish the ACTH dependence.
2) Step 2 (not needed if there is clearly ACTH-independent hypercortisolism): IV CRH 1-microg/kg or IV DDAVP 10-microg stimulation and/or high-dose DST.
a) The IV CRH or DDAVP stimulation test involves stimulation of ACTH secretion and indirect stimulation of cortisol secretion with CRH or DDAVP. This test can be helpful if the equivocal ACTH level is 2.2 to 4.4 pmol/L, where it is not totally clear if there is ACTH dependency or not, since in Cushing disease (which is really ACTH-dependent) ACTH levels increase by ≥35% and cortisol, by ≥20%. In true ACTH-independent Cushing syndrome and in most cases of ectopic ACTH, usually no or minor response to CRH or DDAVP is seen. This test can also help differentiate between pituitary and ectopic sources of ACTH secretion. An increase in the ACTH level by ≥35% and cortisol by 20% is seen with pituitary sources of ACTH, but not with ectopic sources of ACTH.
b) A high-dose dexamethasone 8-mg overnight suppression test is used to differentiate between pituitary and ectopic sources of ACTH production. Patients are instructed to take 8 mg of oral dexamethasone at bedtime (11 pm to midnight). Serum cortisol levels are measured on day one at 8 am and on the following day after dexamethasone administration between 8 and 9 am. Cortisol level suppression >50% is suggestive of pituitary source of abnormal ACTH production, and suppression >90% is even more specific for pituitary source/Cushing disease.
3) Step 3: Imaging studies. Imaging should not be performed until biochemical workup confirms Cushing syndrome and localization based on ACTH tests (above) is completed. This will prevent the risk of finding incidentalomas.
a) Computed tomography (CT) is the first choice for the evaluation of the adrenal gland in ACTH-independent Cushing syndrome. CT helps distinguish between tumors, nodules, and hyperplasia and in most cases can differentiate between adrenal adenomas and carcinomas. Magnetic resonance imaging (MRI) of the adrenal gland is a good option when CT is contraindicated or unavailable.
b) MRI of the sella (Tesla-3) is the test of choice to assess for ACTH-secreting pituitary adenoma (50%-60% sensitivity). The most frequent findings are microadenomas. If there is an adenoma sized >6 mm and concordant biochemical test results (see above), Cushing disease can be diagnosed.
c) Bilateral inferior petrosal sinus sampling (BIPSS): If biochemical test results are discordant or pituitary adenoma is absent on MRI or measures <6 mm, BIPSS performed by an experienced interventional neuroradiologist can be used to differentiate between pituitary and ectopic sources of ACTH.
d) The first imaging choice in identifying ectopic sources of ACTH production is CT of the chest, followed by CT of the abdomen and pelvis.
e) Molecular imaging/receptor scintigraphy with somatostatin analogues (68Ga dotatate positron emission tomography [PET]-CT scan) is used in specialized settings to detect ectopic ACTH-secreting NETs.
f) Skeletal radiographs may reveal features of osteoporosis and pathologic fractures; delayed bone age is frequently seen in children and adolescents. Dual-energy X-ray absorptiometry (DXA) may reveal features of osteopenia or osteoporosis, particularly in the lumbar spine and proximal femur.
CT and MRI of the adrenal glands reveal findings dependent on the cause of Cushing syndrome (Figure 1):
1) Autonomous adrenal cortex tumor or tumors: CT reveals a unilateral adrenal tumor with features of adenoma, atrophy of the contralateral adrenal gland, and less frequently bilateral multiple adenomas of the adrenal cortex. MRI reveals significant fat content and rapid contrast washout.
2) Macronodular adrenal hyperplasia: CT reveals adrenal glands that are usually enlarged and often polycyclic, with large nodules; their density is typical for adenomas. MRI reveals high fat content in the adrenal glands.
3) Micronodular adrenal hyperplasia: CT and MRI usually reveal symmetric adrenal glands that are of normal or slightly increased size, with small nodules. Diagnosis is established during surgery (characteristic yellow-black color of the nodules caused by lipofuscin deposition).
Diagnostic algorithm: Figure 1. Elevated cortisol levels must be found on ≥2 separate test results.
1. Overt Cushing syndrome: Signs and symptoms of Cushing syndrome and evidence of hypercortisolism as above.
2. Subclinical Cushing syndrome due to autonomous adrenal cortisol secretion: Here, suppression with dexamethasone is impaired, while late-night salivary cortisol levels and 24-hour UFC excretion may be normal or may approach or slightly exceed the ULN. The most important diagnostic findings are considered to be the reduction of morning plasma ACTH levels below the lower limit of normal (LLN) and inadequate suppression of cortisol secretion by dexamethasone (serum cortisol levels >50 nmol/L in the overnight suppression test with dexamethasone 1 mg). This type of Cushing syndrome is diagnosed in a small proportion of incidentally detected, usually unilateral, autonomous tumors of the adrenal cortex. A small group of patients who undergo unilateral adrenalectomy for an apparently nonfunctioning adrenal tumor are retrospectively diagnosed with subclinical Cushing syndrome based on postoperative central adrenal insufficiency.
Differential diagnosis of Cushing syndrome: Figure 1.
Other conditions associated with elevated glucocorticoid levels: Glucocorticoid resistance: A very rare syndrome of partially impaired response of the glucocorticoid receptor (a rare genetic condition). It is associated with high serum ACTH, cortisol, androgen, and aldosterone levels without symptoms of excess cortisol, with features of androgenization and aldosteronism in women. The circadian rhythm of cortisol secretion as well as pituitary and adrenal response to CRH are preserved. Treatment: Dexamethasone 1 to 1.5 mg/d to suppress ACTH secretion.
ComplicationsTop
1. Cardiovascular: Hypertension, myocardial infarction, stroke.
2. Metabolic: Diabetes, obesity, hepatic steatosis.
3. Cutaneous: Poor wound healing, thinning.
4. Reproductive: Sexual dysfunction, infertility, androgenism.
5. Thromboembolic: Venous and arterial thromboembolism.
6. Infection: Bacterial, viral, fungal, opportunistic.
7. Neuropsychiatric: Anxiety, depression, psychosis.
8. Musculoskeletal: Osteoporosis, fractures, myopathy.
TreatmentTop
1. Treatment and prevention of complications of Cushing syndrome: Some complications resolve after successful treatment of the underlying conditions that have caused Cushing syndrome.
2. General measures:
1) Blood pressure management: Mineralocorticoid receptor antagonists such as spironolactone can be used, particularly if hypokalemia is present.
2) Diabetes, obesity, coronary artery disease, and hyperlipidemia are to be managed as per relevant society guidelines (see Diabetes; see Obesity; see Coronary Artery Disease; see Hyperlipidemia).
3. Prevention of complications:
1) P jiroveci pneumonia prophylaxis should be considered in prolonged/severe Cushing syndrome or other conditions posing high risk of this infection. Trimethoprim (TMP)/sulfamethoxazole (TMP 80-160 mg/d or 160 mg 3 times/wk) is the first-line treatment and should be continued 2 weeks following curative surgery or normalization of UFC.
2) Venous thromboembolism (VTE) prophylaxis should be considered perioperatively along with active mobilization in high-risk patients.
3) Vaccines: Inactivated (eg, influenza, hepatitis, pneumococcal infections) and mRNA vaccines (eg, severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]) are generally safe. Live attenuated vaccines (eg, measles, mumps, rubella, varicella, herpes zoster) should be avoided in immunocompromised patients or those on immunosuppressants. In patients with Cushing syndrome, a live attenuated vaccine can be given when the disease has been in remission and patients are no longer immunocompromised.
4) Osteoporosis: Given the absence of sufficient evidence, the management of bone mineral disease in Cushing syndrome should be similar to the one used in patients treated with exogenous glucocorticoids. Sufficient calcium and vitamin D intake should be encouraged. The decision on antiresorptive therapy should be made based on overall osteoporosis risk factors using risk calculators. The bone mineral density may be normal in these patients despite high fracture risk.
Treatment of Cortisol Hypersecretion
This depends on the etiology of Cushing syndrome. Usually surgery is the treatment of choice for most causes of Cushing syndrome, but at times medical therapy and radiotherapy may be indicated in addition to other systemic therapies depending on the etiology, clinical circumstances, and patient preference.
An overview of treatments: Table 1.
1. Surgery:
1) Adrenal etiology:
a) Adrenal cortex tumors: The treatment of choice is usually surgical resection of the adrenal tumor.Evidence 1Strong recommendation (benefits clearly outweigh downsides; right action for all or almost all patients). Low Quality of Evidence (low confidence that we know true effects of the intervention). Quality of Evidence lowered due to the observational nature of most studies. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing's syndrome. Lancet. 2015 Aug 29;386(9996):913-27. doi: 10.1016/S0140-6736(14)61375-1. Epub 2015 May 21. Review. PMID: 26004339. After surgical tumor resection, use hydrocortisone replacement (typical starting dose, 30-40 mg/d), as functioning of the contralateral adrenal gland is usually impaired. Over the course of the following weeks, taper the dose of hydrocortisone down to discontinuation. For the following 1 to 2 years, hydrocortisone treatment may be occasionally necessary in the event of severe stress (eg, surgery).
b) Macronodular or micronodular adrenal hyperplasia: The treatment of choice is resection of the largest adrenal gland. Administer and taper hydrocortisone as in patients after resection of a unilateral adenoma.
2) Pituitary etiology (Cushing disease): The first-line treatment is surgical resection of the pituitary tumor (commonly done through the transsphenoidal route).
3) Ectopic ACTH-secreting tumors: Treatment directed at addressing the underlying cause by primary surgical removal of the ectopic sources. Bilateral adrenalectomy can be considered as a curative choice for cortisol excess if primary surgical therapy, medical therapy, or both are not successful or in cases of severe life-threatening Cushing syndrome, where it can be lifesaving.
2. Medical therapy may be indicated to manage hypercortisolism when:
1) The patient is awaiting surgery, especially if it is delayed or the disease is severe.
2) Surgery is contraindicated.
3) Surgery is unsuccessful.
4) Medical therapy is the patient’s preference.Evidence 2Strong recommendation (benefits clearly outweigh downsides; right action for all or almost all patients). Low Quality of Evidence (low confidence that we know true effects of the intervention). Quality of Evidence lowered due to the observational nature of most studies. Nieman LK, Biller BM, Findling JW, et al; Endocrine Society. Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31. doi: 10.1210/jc.2015-1818. Epub 2015 Jul 29. PMID: 26222757; PMCID: PMC4525003.
All of the above are used in specialized settings. Types of medical therapies:
1) Adrenal-directed therapy: There are 2 categories of adrenal-specific therapies:
a) Agents that decrease adrenal cortisol production (eg, ketoconazole 400-1600 mg/d in 2-3 divided doses; metyrapone 500 mg up to 6 g/d in divided doses tid-qid); osilodrostat; or uncommonly mitotane.
b) Agents that block the action of cortisol on adrenal receptors (eg, mifepristone).
2) Pituitary-directed therapy (only for pituitary Cushing/Cushing disease): Somatostatin analogues (eg, pasireotide) and dopamine agonists (eg, cabergoline). The doses of these medications can be titrated to achieve optimal UFC levels.
3) Ectopic ACTH-secreting tumors: Steroidogenesis inhibitors, glucocorticoid receptor antagonists, and somatostatin analogues can be used if surgery is unsuccessful, contraindicated, or both, or while awaiting surgery.
4) Radiation to the pituitary gland (for pituitary Cushing) should be considered if surgery is contraindicated, surgical or medical treatment is unsuccessful, or in cases of tumor recurrence or Nelson syndrome. Both conventional radiation and stereotactic fractionated radiotherapy (using dynamic image–guided radiation) are offered.
Care should be taken to avoid a precipitating glucocorticoid deficit and features of impending adrenal crisis while on any medical therapy. Perioperative administration of glucocorticoids is the same as perioperative management of patients with adrenal insufficiency while on medical therapy that lowers cortisol levels.
PrognosisTop
Regardless of etiology, patients with long-standing Cushing syndrome are at risk of long metabolic and cardiovascular complications that need to be monitored for and addressed.
1. Untreated Cushing syndrome carries a poor prognosis as a result of cardiovascular, thrombotic, and infectious complications.
2. Resection of adrenal adenomas results in complete resolution of physical features of Cushing syndrome. The metabolic complications improve with treatment but may not resolve. Periodic replacement therapy may be needed.
3. Adrenalectomy (often unilateral) in patients with micronodular and macronodular adrenal hyperplasia results in resolution of symptoms of Cushing syndrome. In patients with Carney complex, the prognosis depends on coexisting abnormalities.
4. In patients with adrenocortical carcinoma, the prognosis depends on the stage of cancer, extent of surgery, and response to adjuvant therapy.
5. Cushing disease (pituitary) has a high risk of recurrence and life-long monitoring is indicated.
Tables and FiguresTop
|
Treatment |
Adrenal etiology | Pituitary etiology | Ectopic ACTH secretion |
|
Surgical |
– Adenoma/carcinoma: tumor resection – Macronodular or micronodular adrenal hyperplasia: resection of largest adrenal gland |
– Resection of pituitary tumor – Bilateral adrenalectomy in selected cases |
– Resection of primary tumor – Tumor debulking – Bilateral adrenalectomy in selected cases |
|
Medical |
– Steroidogenesis inhibitors (ketoconazole, metyrapone, osilodrostat, mitotane, etomidate) – Glucocorticoid receptor antagonists (mifepristone) – Chemotherapy for adrenal carcinoma |
– Somatostatin analogues (pasireotide) – Dopamine agonists (cabergoline) – Steroidogenesis inhibitors (ketoconazole, metyrapone, osilodrostat, mitotane, etomidate) – Glucocorticoid receptor antagonists (mifepristone) |
– Steroidogenesis inhibitors (ketoconazole, metyrapone, osilodrostat, mitotane, etomidate) – Glucocorticoid receptor antagonists (mifepristone) – Somatostatin analogues (octreotide/lanreotide) – Everolimus – Tyrosine kinase inhibitors – Chemotherapy |
|
Radiotherapy |
External-beam radiation therapy for adrenal carcinoma |
Conventional versus stereotactic fractionated radiotherapy |
External-beam radiation therapy |
|
Other specialized therapies |
Not applicable |
Not applicable |
– Hepatic artery embolization – Radiofrequency ablation – Peptide-receptor radiolabeled therapy |
|
ACTH, adrenocorticotropic hormone. | |||
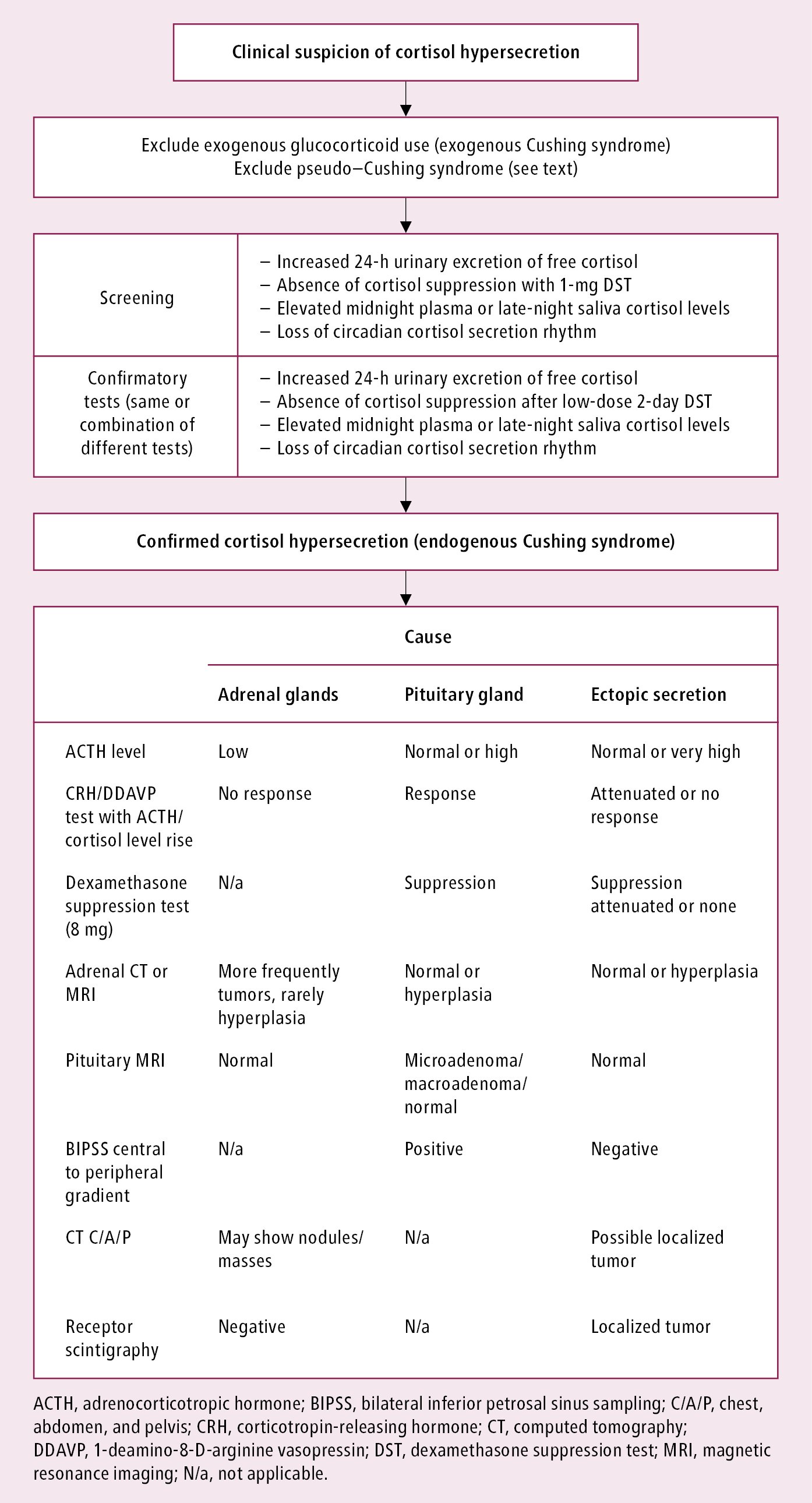
Figure 6.1-1. Diagnostic algorithm in Cushing syndrome.
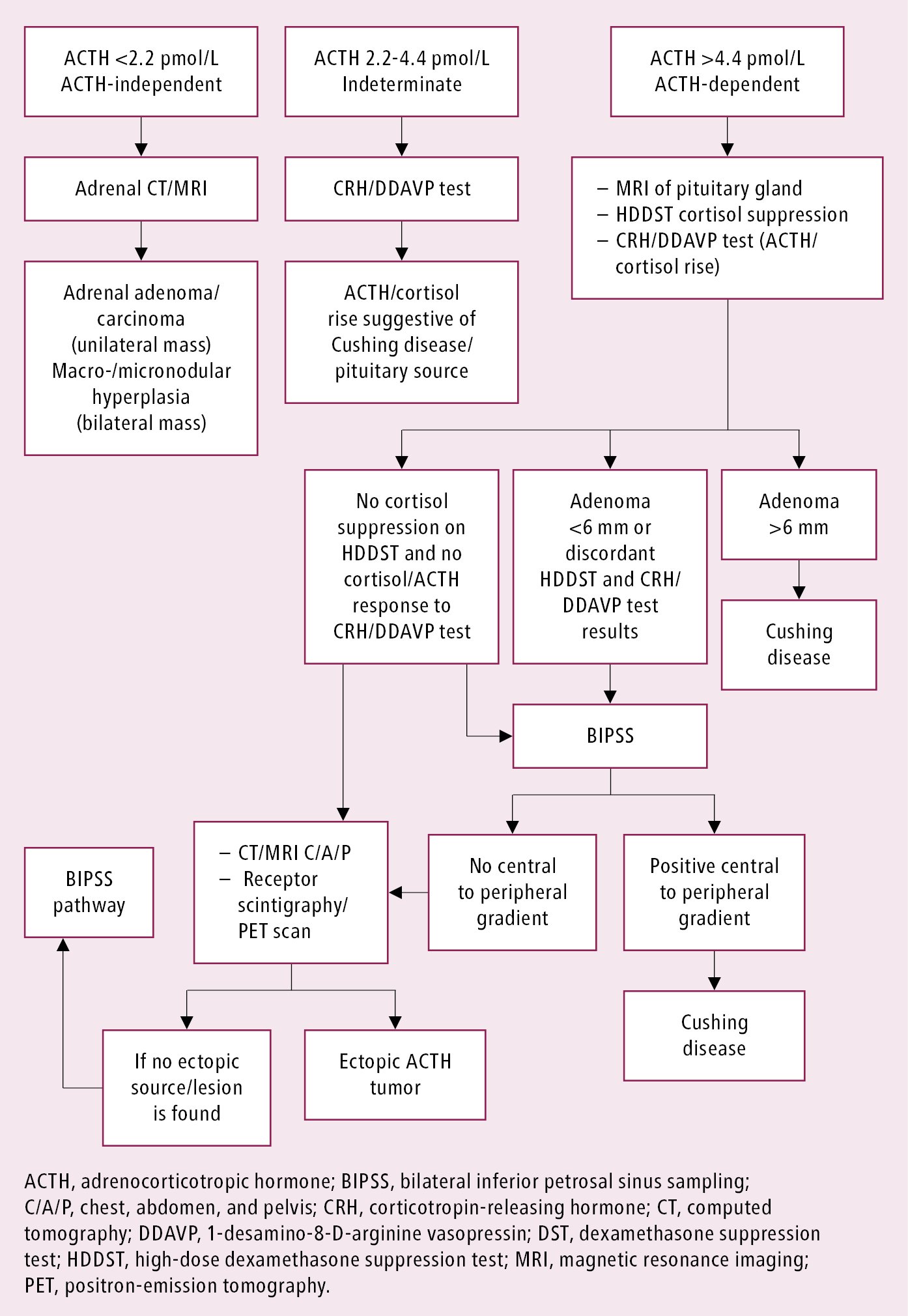
Figure 6.1-2. Approach to identifying the cause of endogenous Cushing syndrome.
 English
English
 Español
Español
 українська
українська