 українська
українська
 English
English
 Español
Español
портал для лікарів
портал для лікарів
ПМ є набутим, ідіопатичним, хронічним міозитом. ДМ є формою міозиту з супутнім дерматитом. При обидвох захворюваннях можуть розвинутись запальні зміни в серці, інтерстиціальній тканині легень та кровоносних судинах.
Етіологія невідома. Вважають, що головну роль у патогенезі ПМ/ДМ відіграють аутоімунні механізми.
КЛІНІЧНА КАРТИНА ТА ТИПОВИЙ ПЕРЕБІГ
ПМ і ДМ належать до найчастіших ідіопатичних запальних міопатій у дорослих. Жінки хворіють у 2 рази частіше, ніж чоловіки. Хвороба може розвинутись у будь-якому віці, пік захворюваності припадає на вік 10–15 років (дитяча форма) i 35–65 років. Початок захворювання може бути гострим (декілька днів), підгострим (тижні) або хронічним (місяці, роки). У більшості нелікованих хворих розвивається повільна атрофія м'язів та їх контрактура. 5-річна смертність ≈50 %. ДМ підвищує ймовірність наявності злоякісної пухлини у 6 разів, а ПМ — у 2 рази (підвищений ризик раків: яєчника, молочної залози, легені, шлунка, кишківника, порожнини носа і горла, підшлункової залози, сечового міхура та неходжкінських лімфом).
1. Загальні симптоми: слабкість, підвищення температури тіла, втрата маси тіла.
2. Симптоми ураження м'язів:
1) переважно симетрична слабкість у м’язах плечового поясу, тазового поясу, а також м’язів потилиці та спини (майже у всіх хворих), призводить до труднощів зі вставанням із сидячого положення, підніманням важких предметів, причісуванням волосся, ходьбою по сходах і т. д., м'язи часто стають чутливими та болючими;
2) слабкість дихальних м'язів — викликає дихальну недостатність;
3) ослаблення м'язів горла, стравоходу та гортані — призводить до дисфагії та дисфонії;
4) ураження м'язів очного яблука (рідко) — ністагм, погіршення зору.
3. Дерматологічні зміни: виявляються при ДМ; їх поява і вираженість не завжди пов'язані з ураженням м'язів; можуть випереджати міозит або розвиватись самостійно (CADM, dermatomyositis sine myositide [дерматоміозит без міозиту]). Еритематозні зміни часто супроводжуються свербіжем і/або фотосенсибілізацією.
1) еритема навколо очей у формі окулярів (рис. 16.6-1), з ліловим забарвленням (так звана геліотропна), іноді з набряком повік — патогномонічний симптом, спостерігається у 30–60 % хворих; еритема декольте у формі літери V; додатково еритема задньої частини шиї і плечей (симптом шалика), бічної поверхні стегон (симптом кобури);
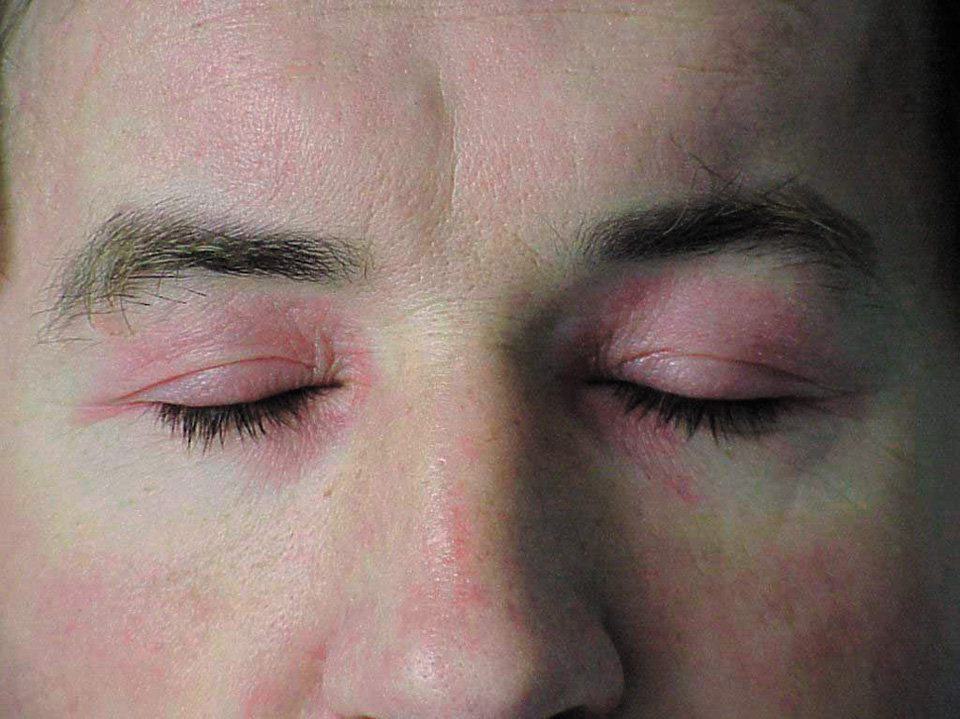
Рисунок 1. ПМ/ДМ — геліотропна еритема навколо очей
2) папули Готтрона — папули синюшного відтінку з гіпертрофією епідермісу, що розташовані на розгинальній поверхні суглобів (найчастіше міжфалангових і п'ястно-фалангових суглобів →розд. 32.1; іноді суглобів кистей, ліктьових, колінних та гомілково-стопних суглобів; симптом Готтрона це еритематозні або синюшного відтінку плями в тій самій локалізації (патогномонічні симптоми; у ≈70 % хворих);
3) інші — огрубіння, лущення і утворення тріщин на подушечках пальців та долонній поверхні кистей (т. зв. руки механіка, рідко); на нігтьових валиках еритема з набряком, петехіями і телеангіектазіями; трофічні виразки внаслідок супутнього шкірного васкуліту; генералізована еритродермія; панікуліт; сітчасте ліведо; вогнищева алопеція без утворення рубців.
4. Ураження серця: у 70 %, зазвичай тахікардія або брадикардія, рідко прояви серцевої недостатності.
5. Ураження легень: симптоми інтерстиціальної хвороби легень (у 30–40 %), головним чином сухий кашель і наростаюча задишка, з часом хронічна дихальна недостатність; аспіраційна пневмонія у хворих із дисфагією.
6. Ураження ШКТ: симптоми порушень моторики стравоходу, шлунка і кишківника, в т. ч. гастроезофагального рефлюксу; у тяжких випадках виразки і кровотечі.
7. Ураження суглобів: симптоми неерозивного артриту або біль у суглобах, особливо периферичних, переважно у суглобах кистей (у 20–50 %).
8. Кальцинати — у підшкірній тканині, скелетних м'язах, фасціях і сухожиллях (у >10 %), іноді масивні.
9. Синдром Рейно (у 10–15 %).
1. Лабораторні дослідження
1) дослідження крові — підвищення активності м'язевих ферментів у сироватці — КФК, АСТ, АЛТ, ЛДГ, альдолази (активність у межах норми не виключає ПМ/ДМ), підвищення ШОЕ, підвищення концентрації міоглобіну, СРБ і гаммаглобулінів у сироватці;
2) імунологічні дослідження — ANA (у 40–80 %), у т. ч. антитіла, що асоційовані з ПМ/ДМ (до аміноацил-тРНК-синтетаз [найчастіше, анти-Jo-1], анти-SRP i анти-Mi-2) i супутні антитіла (анти-Ro, анти-La, анти-U1-RNP, анти-U2-RNP, анти-Ku, анти-Sm, анти-PM/Scl).
2. Електроміографія: виявляє ознаки первинного ушкодження м'язів.
3. Гістологічні дослідження:
1) біоптат м'язу — запальна інфільтрація в основному лімфоцитами; тип клітин, структури, втягнуті в запальний процес, і наявність періфасцикулярної атрофії залежать від форми захворювання;
2) біоптат легені (хірургічна біопсія) — може виявити інтерстиціальну хворобу легень (різні форми).
4. Візуалізаційні дослідження: МРТ м'язів — підсилення сигналу в Т1- i Т2-залежних зображеннях; Т2-залежні зображення із супресією жирів або у STIR-зваженому режимі характеризуються найвищою чутливістю для виявлення активного запалення, однак виявлені зміни не є патогномонічними для ПМ/ДМ; дослідження може полегшити вибір оптимального місця для забору м'язевого біоптату. РГ і КТВР грудної клітки — інтерстиціальні зміни. РГ кісток і суглобів — може виявити множинні кальцинати, переважно в підшкірній тканині та м'язах, а також остеопороз.
5. ЕКГ: зазвичай неспецифічні зміни інтервалу ST і зубця T, синусова тахікардія, рідше — АВ-блокада I або II ступеню.
На основі діагностичних критеріїв →табл. 16.6-1.
|
критерії за Alarcon-Segovia i Villareal |
|
1) серологічні: наявність антитіл anty-U1 RNP у титрі ≥1:1600 2) клінічні: a) набряк кистей рук б) артрит в) міозит г) синдром Рейно д) склеродактилія |
|
діагноз ЗЗСТ = відповідність серологічному критерію та ≥3-м клінічним критеріям (у разі співіснування набряку кистей рук, синдрому Рейно і склеродактилії додатково вимагається відповідність критерію 2б або 2в) |
|
критерії за Kasukawa та співавт. |
|
I. спільні симптоми: 1) синдром Рейно 2) набряк пальців або кистей рук II. наявність антитіл анти-U1 RNP III. «змішані» симптоми A. симптоми, що нагадують СЧВ: 1) поліартрит 2) лімфаденопатія 3) еритема обличчя 4) перикардит або плеврит 5) лейкопенія або тромбоцитопенія Б. симптоми, що нагадують системну склеродермію: 1) склеродактилія 2) фіброз легень, рестриктивні зміни або знижена TLCO 3) ослаблена перистальтика або розширення стравоходу В. симптоми, що нагадують ПМ: 1) слабкість м'язів 2) підвищення КФК у сироватці крові 3) ознаки міогенного механізму ушкодження м'язів на ЕМГ |
|
діагноз ЗЗСТ = наявність ≥1 спільного (I) симптому та серологічного (II) критерію, а також ≥1 «змішаного» симптому з кожної групи (A, Б, В) |
|
критерії за Танакі та співавт. |
|
I. загальні симптоми — як за Касукавою II. наявність анти-U1 RNP антитіл III. ураження характерних органів: 1) легенева артеріальна гіпертензія; 2) стерильний менінгіт; 3) невропатія трійчастого нерва IV. «змішані» симптомиA. симптоми, схожі з СЧВ: 1) поліартрит; 2) збільшені лімфатичні вузли; 3) еритема обличчя; 4) перикардит або плеврит; 5) лейкопенія (≤4000/мкл) або тромбоцитопенія (≤100 000/мкл) Б. симптоми, подібні до СС: 1) склеродактилія; 2) інтерстиціальна хвороба легень; 3) ослаблення перистальтики або розширення стравоходу В. симптоми, подібні до ПМ/ДМ: 1) м’язова слабкість; 2) підвищення активності м'язових ферментів у сироватці крові; 3) особливості міогенного ураження м'язів при ЕМГ |
|
діагноз ЗЗСТ: ≥1 симптом із загальним (I), серологічним критерієм (II) та ≥1 симптомом ураження характерного органу (III) або «змішаними» симптомами (IV) — ≥1 із ≥2 груп (А, Б або В) |
1) ідіопатичні запальні міопатії, інші, ніж ПМ/ДМ — зазвичай міозит, супутній до інших системних захворювань сполучної тканини (оверлеп-синдроми), міозит, асоційований з онкозахворюванням, аутоімунна некротична міопатія (клінічно відповідає ПМ, часто пов’язана з системним захворюванням сполучної тканини, вірусною інфекцією (напр., ВІЛ), антитілами анти-SRP, прийомом статинів або онкозахворюванням; при гістологічному дослідженні характеризується відсутністю типового інфільтрату одноядерних клітин, зазвичай дає добру відповідь на імуносупресивні ЛЗ); міозит із внутрішньоклітинними включеннями (домінує ослаблення дистальних м'язів, іноді несиметричне, без шкірних змін); міозит із еозинофілією, вогнищевий міозит (болюча припухлість, найчастіше розташована на нижній кінцівці, початок переважно гострий, без травми в анамнезі, у 90 % минає без лікування); запалення орбітальних м'язів;
2) патологічні стани з вторинним ушкодженням або слабкістю м'язів →табл. 16.6-2. Необхідно шукати злоякісну пухлину.
|
Маніфестацїі |
ССД |
РА |
СЧВ |
СШ |
ЗЗСТ |
ПM/ДМ |
|
неспецифічна інтерстиціальна пневмонія |
+++ |
++ |
++ |
++ |
++ |
++ |
|
звичайна інтерстиціальна пневмонія (ЗІП) |
+++ |
++ |
+ |
+ |
++ |
++ |
|
криптогенна організуюча пневмонія |
+ |
++ |
+ |
+ |
|
++ |
|
лімфоцитарна інтерстиціальна пневмонія |
|
+ |
+ |
+ |
|
|
|
гострий вовчаковий пневмоніт |
|
|
+ |
|
|
|
|
гостра інтерстиціальна пневмонія |
|
|
|
|
|
+ |
|
аспіраційна пневмонія |
+ |
|
|
|
|
+ |
|
легеневі вузлики |
|
+ |
|
|
|
|
|
облітеруючий бронхіоліт |
|
+ |
|
+ |
|
+ |
|
фолікулярний бронхіоліт |
|
+ |
|
+ |
|
|
|
інші форми бронхіолітів |
|
|
|
+ |
|
|
|
бронхоектази |
|
++ |
|
+ |
|
|
|
ксеротрахея |
|
|
|
+ |
|
|
|
васкуліти |
|
+ |
+ |
|
|
+ |
|
легенева гіпертензія |
+ |
|
+ |
|
+ |
|
|
антифосфоліпідний синдром |
|
|
+ |
|
|
|
|
дифузна альвеолярна кровотеча |
|
|
+ |
|
|
+ |
|
ураження плеври |
|
+++ |
+++ |
|
++ |
|
|
екзогенна рестрикція |
+ |
|
|
|
|
|
|
слабкість дихальних м'язів |
|
|
|
|
|
+ |
|
синдром «зморщеної легені» |
|
|
+ |
|
|
|
|
а Пропущено васкуліти, не пов'язані з системними захворюваннями, наведеними в таблиці. ЗЗСТ — змішане захворювання сполучної тканини, ПM/ДМ — поліміозит і дерматоміозит, РА — ревматоїдний артрит, СЧВ — системний червоний вовчак, СШ — синдром Шегрена, ССД — системна склеродермія | ||||||
1. ГК: преднізон п/о 1 мг/кг/добу; гострий початок або тяжчий перебіг → при початковому лікуванні можна застосувати в/в метилпреднізолон 0,5–1,0 г протягом 3 днів. Після покращення стану (зростання м'язевої сили, нормалізація маркерів ушкодження м'язів), але не раніше, ніж через 4–8 тиж. після початку лікування, слід поступово знижувати добову дозу перорального ГК, напр., на ≈10 мг на місяць, до підтримуючої дози 5–10 мг/добу і продовжувати лікування протягом кількох років, а іноді — до кінця життя. Деякі автори рекомендують знижувати дозу на 5 мг кожного тижня до 20 мг/добу, в подальшому на 2,5 мг кожні 2 тижні до 10 мг/добу, потім на 1 мг кожного місяця до відміни ЛЗ включно.
2. Якщо впродовж 6 тиж. від початку лікування не наступило задовільне покращення, або якщо перебіг захворювання є блискавичним → додайте один із ЛЗ (препарати →табл. 16.1-6): метотрексат п/о або парентерально (в/м або п/ш) 1 × на тиж., з фолієвою або фоліновою кислотою; починайте, в залежності від тяжкості захворювання, від 10–15 мг 1 × на тиж., якщо симптоми захворювання не минули, поступово збільшуйте на 5 мг кожних 2–3 міс. або швидше; азатіоприн 1,5–2 мг/кг/добу, дозу знижуйте на 25 мг кожного місяця, до підтримуючої дози 50 мг/добу; терапевтичний ефект проявляється через >3 міс.; циклоспорин 2,5–5 мг/кг/добу; циклофосфамід 1,0 г в/в 1 × на міс., у разі потреби — 1–3 мг/кг 1 × на день п/о — застосовується переважно у разі інтерстиціальних змін у легенях або вираженого васкуліту; мофетил мікофенолату 2–3 г/добу — у разі неефективності або непереносимості метотрексату і циклоспорину; гідроксихлорохін 200 мг 2 × на день або хлорохін 250 мг 2 × на день — показані у разі резистентних дерматологічних змін; ВВІГ — показані у разі неефективності імуносупресивного лікування; 0,4 г/кг/добу протягом 5 наступних днів 1 × на міс. впродовж 3–7 наступних місяців; ритуксимаб 750 мг/м2 пт. (макс. 1 г) 2 рази з інтервалом в 2 тиж. У випадках резистентності до іншого імуносупресивного лікування тривають спроби застосування такролімусу, препаратів анти-ФНП.
У процесі відновлення функціональної здатності важливою є кінезотерапія. У гострій фазі захворювання в основному слід застосовувати пасивні вправи, які запобігають виникненню контрактур; під час реконвалесценції — ізометричні вправи і вправи для розвантаження, пізніше додатково ізотонічні і аеробні вправи; дуже корисними є вправи у воді. Фізичне навантаження у цей період не повинно перевищувати 60 % максимального використання кисню.
Періодична оцінка м'язевої сили, активності КФК у сироватці, ЕМГ, а у хворих з інтерстиціальним захворюванням легень — КТВР, функціональні дослідження легень і тест з 6-хвилинною ходьбою. Важливо є зберігати онкологічну настороженість з огляду на підвищений ризик розвитку злоякісних пухлин, який є максимальним протягом перших 3 років від встановлення діагнозу ПМ/ДМ, однак зберігається навіть через 5 років. Ризик розвитку неоплазії підвищують з-поміж іншого вік >50 років, ДМ, тяжкі дерматологічні зміни, наявність антитіл анти-155/140 кД, відсутність супутніх або асоційованих з ПМ/ДМ антитіл.
У разі правильного лікування 10 років переживуть >80 %. Прогноз погіршують в т. ч. старший вік і ураження внутрішніх органів, особливо легень, наявність супутньої злоякісної пухлини, наявність антитіл анти-SRP.



