 українська
українська
 English
English
 Español
Español
портал для лікарів
портал для лікарів
Ми прагнемо й надалі безкоштовно надавати цей тип контенту. На жаль, коштів на це більше немає.
Без Вашої допомоги нам доведеться закрити проект до кінця 2024 року.
Зробіть пожертвуВИЗНАЧЕННЯ ТА ЕТІОПАТОГЕНЕЗ вгору
Поліміозит (ПМ; polymyositis — PM) — є набутим, ідіопатичним, хронічним міозитом. Дерматоміозит (ДМ; dermatomyositis — DM) є формою міозиту з супутнім дерматитом. При обидвох захворюваннях можуть розвинутись запальні зміни в серці, інтерстиціальній тканині легень та кровоносних судинах.
Етіологія невідома. Вважають, що головну роль у патогенезі ПМ/ДМ відіграють аутоімунні механізми.
Аутоімунна некротична міопатія може бути пов’язана із застосуванням статинів, вірусною інфекцією, антитілами анти-SRP або анти-HMGCR та онкологічним захворюванням.
КЛІНІЧНА КАРТИНА ТА ТИПОВИЙ ПЕРЕБІГвгору
ПМ і ДМ належать до найчастіших ідіопатичних запальних міопатій у дорослих. Жінки хворіють у 2 рази частіше, ніж чоловіки. Хвороба може розвинутись у будь-якому віці, пік захворюваності припадає на вік 10–15 років (дитяча форма) i 35–65 років. Початок захворювання може бути гострим (декілька днів), підгострим (тижні) або хронічним (місяці, роки). У більшості нелікованих пацієнтів розвивається повільна атрофія м’язів та їх контрактура. 5-річна смертність ≈50 %. ДМ підвищує ймовірність наявності злоякісної пухлини у 6 разів, а ПМ — у 2 рази (підвищений ризик раків: яєчника, молочної залози, легень, шлунка, кишківника, порожнини носа і горла, підшлункової залози, сечового міхура та неходжкінських лімфом).
1. Загальні симптоми: слабкість, підвищення температури тіла, втрата маси тіла.
2. Симптоми ураження м’язів:
1) переважно симетрична слабкість у м’язах плечового поясу, тазового поясу, а також м’язів потилиці та спини (майже у всіх хворих), призводить до труднощів зі вставанням із сидячого положення, підніманням тяжких предметів, причісуванням волосся, ходьбою по сходах і т. д., м’язи часто болючі і стають пальпаторно чутливими;
2) слабкість дихальних м’язів — викликає дихальну недостатність;
3) ослаблення м’язів горла, стравоходу та гортані — призводить до дисфагії та дисфонії;
4) ураження м’язів очного яблука (рідко) — ністагм, погіршення зору.
3. Дерматологічні зміни: виявляються при ДМ; їх поява і вираженість не завжди пов’язані з ураженням м’язів; можуть випереджати міозит або розвиватись самостійно (CADM, dermatomyositis sine myositide [дерматоміозит без міозиту]). Еритематозні зміни часто супроводжуються свербежем і/або фотосенсибілізацією.
1) еритема навколо очей у формі окулярів (→рис. 16.6-1), з ліловим забарвленням (так звана геліотропна), іноді з набряком повік — патогномонічний симптом, спостерігається у 30–60 % пацієнтів; еритема декольте у формі літери V; додатково еритема задньої частини шиї і плечей (симптом шалика), бічної поверхні стегон (симптом кобури);
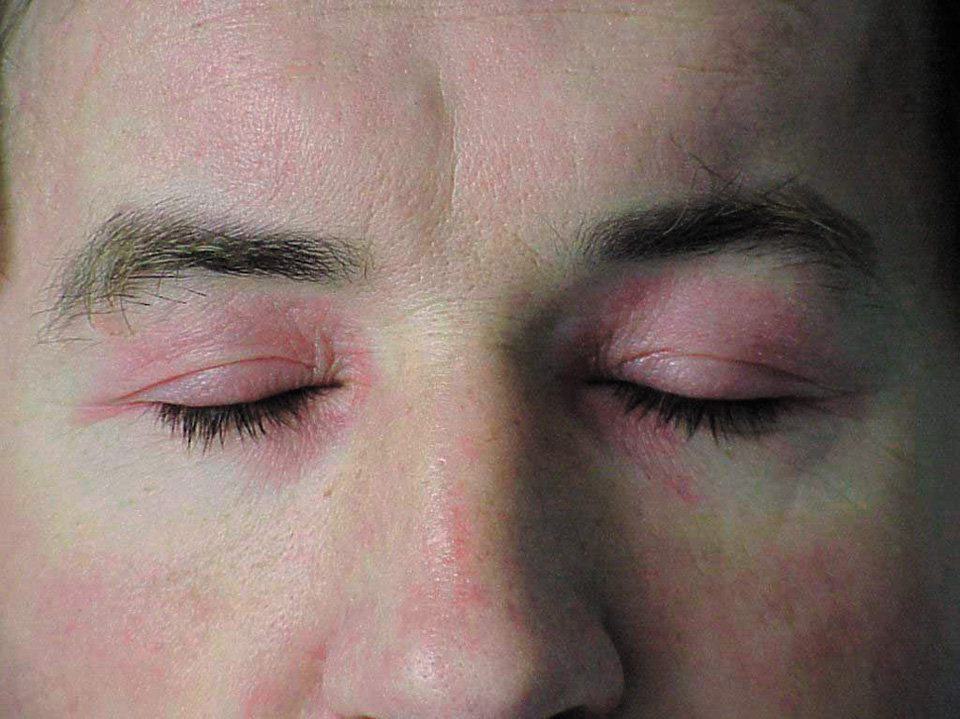
Рисунок 1. ПМ/ДМ — геліотропна еритема навколо очей
2) папули Готтрона — папули синюшного відтінку з гіпертрофією епідермісу, що розташовані на розгинальній поверхні суглобів (найчастіше міжфалангових і п’ястно-фалангових суглобів →рис. 1.31.5; іноді суглобів кистей, ліктьових, колінних та гомілково-стопних суглобів; симптом Готтрона — це еритематозні або синюшного відтінку плями в тій самій локалізації (патогномонічні симптоми; у ≈70 % пацієнтів);
3) інші — огрубіння, лущення і утворення тріщин на подушечках пальців та долонній поверхні кистей (т. зв. руки механіка, рідко); на нігтьових валиках еритема з набряком, петехіями і телеангіектазіями; трофічні виразки внаслідок супутнього шкірного васкуліту; генералізована еритродермія; панікуліт; сітчасте ліведо; вогнищева алопеція без утворення рубців.
4. Ураження серця: у 70 %, зазвичай тахікардія або брадикардія, рідко прояви серцевої недостатності.
5. Ураження легень: симптоми інтерстиціальної хвороби легень (у 30–40 %), головним чином сухий кашель і наростаюча задишка, з часом хронічна дихальна недостатність; аспіраційна пневмонія у пацієнтів із дисфагією.
6. Ураження ШКТ: симптоми порушень моторики стравоходу, шлунка і кишківника, в т. ч. гастроезофагального рефлюксу; в тяжких випадках виразки і кровотечі.
7. Ураження суглобів: симптоми неерозивного артриту або біль у суглобах, особливо периферичних, переважно у суглобах кистей (у 20–50 %).
8. Кальцинати — у підшкірній тканині, скелетних м’язах, фасціях і сухожиллях (у >10 %), іноді масивні.
9. Феномен Рейно (у 10–15 %).
ДІАГНОСТИКАвгору
1. Лабораторні дослідження:
1) дослідження крові — підвищення активності м'язових ферментів у сироватці — КФК, АСТ, АЛТ, ЛДГ, альдолази (активність у межах норми не виключає ПМ/ДМ), підвищення ШОЕ, підвищення концентрації міоглобіну, СРБ і гаммаглобулінів у сироватці;
2) імунологічні дослідження — AНA (у 40–80 %), у т. ч. антитіла, що асоційовані з ПМ/ДМ (до аміноацил-тРНК-синтетаз [найчастіше, анти-Jo-1], анти-SRP i анти-Mi-2) i супутні антитіла (анти-Ro, анти-La, анти-U1-RNP, анти-U2-RNP, анти-Ku, анти-Sm, анти-PM/Scl). Антитіла анти-MDA5 асоціюються з тяжким перебігом уражень шкіри, появою виразок та тяжким перебігом інтерстиціального захворювання легень, зате з відносно низькою активністю самого міозиту. Антитіла анти-SAE з’являються в пацієнтів з ДМ із обширним ураженням м’язів (включаючи дисфагію). Антитіла анти-TIF1γ і NXP2 частіше зустрічаються в пацієнтів з ДМ, асоційований із онкологічним захворюванням.
2. Електроміографія: виявляє ознаки первинного ушкодження м’язів.
3. Гістологічні дослідження:
1) біоптат м’яза — запальна інфільтрація в основному лімфоцитами; тип клітин, структури, втягнуті в запальний процес, і наявність періфасцикулярної атрофії залежать від форми захворювання;
2) біоптат легені (хірургічна біопсія) — може виявити інтерстиціальну хворобу легень (різні форми).
4. Візуалізаційні дослідження: МРТ м’язів — підсилення сигналу в Т1- i Т2- зважених зображеннях; Т2-зважені зображення із супресією жирів або у STIR-зваженому режимі характеризуються найвищою чутливістю для виявлення активного запалення, однак виявлені зміни не є патогномонічними для ПМ/ДМ; дослідження може полегшити вибір оптимального місця для забору м’язового біоптату. Х-променеві дослідження і КТВР грудної клітки — інтерстиціальні зміни. Х-променеві дослідження кісток і суглобів — можуть виявити множинні кальцинати, переважно в підшкірній тканині та м’язах, а також остеопороз.
5. ЕКГ: зазвичай неспецифічні зміни інтервалу ST і зубця T, синусова тахікардія, рідше — АВ-блокада I або II ступеня.
6. Капіляроскопія: більшість пацієнтів з ЦД мають зміни, подібні до системної склеродермії.
Діагноз встановлюється на основі клінічних ознак, особливо м’язової слабкості з типовою локалізацією та перебігом, а також наявності типових уражень шкіри в поєднанні з результатами гістологічного дослідження м’язів та електроміографії. Діагностичні критерії Bohana i Petera можуть бути корисними →табл. 16.6-1. Класифікаційні критерії запальних міопатій за EULAR/ACR →табл. 16.6-2.
|
1) симетрична, наростаюча слабкість м’язів плечового поясу і тазового поясу 2) типова для міозиту гістологічна картина 3) підвищена активність КФК або альдолази в сироватці крові 4) електроміографічні ознаки первинного ушкодження м’язів |
|||||
|
діагноз |
підтверджений |
вірогідний |
можливий |
||
|
кількість симптомів |
|||||
|
ПМ |
4 |
3 |
2 |
||
|
ДМ |
3 або 4 |
2 |
1 |
||
|
та характерні ураження шкіриа |
|||||
|
a Симптом Готтрона, геліотропна еритема повік, еритема декольте або плечей |
|||||
|
Ознака/симптом |
Приклад (визначення) |
Бали |
|
|
з біопсією |
без біопсії |
||
|
вік появи перших симптомів |
≥18 років i <40 років |
1,5 |
1,3 |
|
≥40 років |
2,2 |
2,1 |
|
|
м’язова слабкість |
– слабкість проксимальних м’язів верхніх кінцівок (симетрична, зазвичай прогресуюча)a |
0,7 |
0,7 |
|
– слабкість проксимальних м’язів нижніх кінцівок (симетрична, зазвичай прогресуюча)a |
0,5 |
0,8 |
|
|
– згиначі шиї відносно слабші, ніж розгиначіa |
1,6 |
1,9 |
|
|
– проксимальні м’язи нижніх кінцівок відносно слабші, ніж дистальні м’язиa |
1,2 |
1,9 |
|
|
шкірні симптоми |
– геліотропна еритемаб (→рис. 16.6-1) |
3,2 |
3,1 |
|
– вузлики Готтронав |
2,7 |
2,1 |
|
|
– симптом Готтронаг |
3,7 |
3,3 |
|
|
інші клінічні симптоми |
порушення ковтання або моторики стравоходу |
0,6 |
0,7 |
|
лабораторні дослідження |
– анти-Jo-1 антитіла (позитивний результат тесту, виконаного стандартизованим і прийнятним методом) |
3,8 |
3,9 |
|
– підвищення активності в сироватці крові КФK, АСТ, АЛТ, ЛДГ (найвище значення в ході захворювання) |
1,4 |
1,3 |
|
|
біопсія м’яза |
– інфільтрація перимізію мононуклеарними клітинами, що оточують м’язові волокна, але не проникають у них |
1,7 |
– |
|
– інфільтрація епімізію і/або судинних пучків (в епімізії або перимізії) мононуклеарами |
1,2 |
– |
|
|
– перифасцикулярна атрофія (клітини в декількох рядах навколо судинно-нервових пучків явно менші, ніж клітини, розташовані більш центрально) |
1,2 |
– |
|
|
– вакуолі з облямованими краями (вони забарвлюються в синій колір при фарбуванні гематоксилін-еозином і в червоний колір при модифікованому фарбуванні методом Гоморі) |
3,1 |
– |
|
|
a під час фізикального огляду чи іншого вимірювання б еритема фіолетового відтінку навколо очей, іноді з набряком повік в Eритематозні або синюшні папули з епідермальною гіперплазією, розташовані на розгинальній поверхні суглобів, найчастіше міжфалангових і п’ястно-фалангових, кистей рук; вони також можуть виникати навколо зап’ясть, ліктьових, колінних і надп’ятково-гомілкових суглобів. г еритематозні або синюшні плями в тому ж місці, що й папули Примітка: класифікаційні критерії можна використовувати, коли немає інших причин, що пояснюють симптоми. Діагноз вважається точним (імовірність ≥90 %), якщо результат становить ≥8,7 при біопсії або ≥7,5 без біопсії, та ймовірним (≥55 %), якщо результат відповідно складає ≥6,7 і ≥5,5. АЛТ — аланінамінотрансфераза, АСТ — аспартатамінотрансфераза, КФК — креатинкіназа, ЛДГ — лактатдегідрогеназа на основі: Arthritis Rheum., 2017; 69: 2271–2282; модифіковано |
|||
1) ідіопатичні запальні міопатії, інші, ніж ПМ/ДМ — зазвичай міозит, супутній до інших системних захворювань сполучної тканини (оверлеп-синдроми), міозит, асоційований з онкозахворюванням, аутоімунна некротична міопатія (клінічно нагадує дуже тяжкий ПМ, швидкий розвиток симптомів, часто пов’язана з системним захворюванням сполучної тканини, вірусною інфекцією [напр., ВІЛ], антитілами анти-SRP, прийомом статинів або онкозахворюванням; при гістологічному дослідженні характеризується відсутністю типового інфільтрату одноядерних клітин, зазвичай дає добру відповідь на імуносупресивні ЛЗ); міозит із внутрішньоклітинними включеннями (домінує ослаблення дистальних м’язів, іноді несиметричне, без шкірних змін, у частини пацієнтів наявні специфічні антитіла [анти-cN1A] та накопичення білка TDP-43 в саркоплазмі м’язових волокон); міозит із еозинофілією, вогнищевий міозит (болюча припухлість, найчастіше розташована на нижній кінцівці, початок переважно гострий, без травми в анамнезі, у 90 % минає без лікування); запалення орбітальних м’язів;
2) патологічні стани з вторинним ушкодженням або слабкістю м’язів →табл. 16.6-3.
|
захворювання ендокринної системи |
синдром Іценка-Кушинга, гіпо- і гіпертиреоз, гіпер- і гіпопаратиреоз |
|
метаболічні порушення |
дефіцит карнітину, неправильний метаболізм вітаміну А, глікогенози |
|
захворювання нервової системи |
генетично детерміновані м’язові дистрофії, проксимальна нейропатія, синдром Ламберта-Ітона, міастенія, бічний аміотрофічний склероз |
|
інфекції |
вірусні, бактеріальні, паразитарні (токсоплазмоз, трихінельоз) |
|
порушення електролітного балансу |
знижений або підвищений рівень в плазмі: натрію, калію, кальцію, магнію або фосфатів |
|
гранулематозні захворювання |
саркоїдоз |
|
ЛЗ |
аміодарон, хінідин, циметидин, циклоспорин, даназол, D-пеніциламін, α-інтерферон, інтерлейкін-2, колхіцин, ε-амінокапронова кислота, фенілбутазон, фенітоїн, ГК, гідралазин, карнітин, ліпідознижуючі ЛЗ (фібрати, статини, нікотинова кислота), протималярійні ЛЗ (хлорохін і гідроксихлорохін), леводопа, пеніцилін, прокаїнамід, ріфампіцин, сульфонаміди, L-триптофан, вальпроєва кислота, вінкрістин, зидовудин |
|
токсичні речовини |
оксид вуглецю, алкоголь, героїн, кокаїн |
|
фізичні фактори |
електрошок, травма, гіпертермія |
ЛІКУВАННЯвгору
1. ГК: преднізон п/о 1 мг/кг/добу (макс. 80 мг/кг); гострий початок або тяжчий перебіг → при початковому лікуванні можна застосувати в/в метилпреднізолон 0,5–1,0 г протягом 3 днів. Після покращення стану (нормалізація маркерів ушкодження м’язів, збільшення м’язової сили), але не раніше, ніж через 4–8 тиж. після початку лікування, слід поступово знижувати добову дозу перорального ГК, напр., на ≈10 мг на місяць, до підтримуючої дози 5–10 мг/добу і продовжувати лікування протягом кількох років, а іноді — до кінця життя. Деякі автори рекомендують знижувати дозу на 5 мг кожного тижня до 20 мг/добу, в подальшому на 2,5 мг кожні 2 тижні до 10 мг/добу, потім на 1 мг кожного місяця до відміни ЛЗ включно.
2. Якщо впродовж 6 тиж. від початку лікування не наступило задовільне покращення, або якщо перебіг захворювання є блискавичним, або форма захворювання пов’язана з гіршим прогнозом → додайте один із ЛЗ (препарати →табл. 16.1-6): метотрексат п/о або парентерально (в/м або п/ш) 1 × на тиж., з фолієвою або фоліновою кислотою; починайте, в залежності від тяжкості захворювання, від 10–15 мг 1 × на тиж., якщо симптоми захворювання не минули, поступово збільшуйте на 5 мг кожних 2–3 міс. або швидше; азатіоприн 1,5–2 мг/кг/добу, дозу знижуйте на 25 мг кожного місяця, до підтримуючої дози 50 мг/добу; терапевтичний ефект проявляється через >3 міс.; мофетил мікофенолату 2–3 г/добу — у разі неефективності або непереносимості метотрексату і циклоспорину; гідроксихлорохін 200 мг 2 × на день або хлорохін 250 мг 2 × на день — показані у разі резистентних дерматологічних змін; ВВІГ — показані у разі неефективності імуносупресивного лікування; 0,4 г/кг/добу протягом 5 наступних днів 1 × на міс. впродовж 3–7 наступних місяців; ритуксимаб 750 мг/м2 п. т. (макс. 1 г) 2 рази з інтервалом в 2 тиж; циклоспорин 2,5–5 мг/кг/добу; циклофосфамід 1,0 г в/в 1 × на міс., у разі потреби — 1–3 мг/кг 1 × на день п/о — застосовується переважно у разі інтерстиціальних змін у легенях або вираженого васкуліту. У випадках резистентності до іншого імуносупресивного лікування тривають спроби застосування такролімусу, препаратів анти-ФНП, в/в імуноглобулінів у імуномодулюючих дозах.
3. Лікування інтерстиціального захворювання легень: лікування першої лінії (ACR 2023): мікофенолату мофетил, азатіоприн, ритуксимаб або інгібітор кальциневрину, а додаткові варіанти включають циклофосфамід або інгібітор JAK. Розгляньте короткострокове додавання кортикостероїдів до будь-якого з перерахованих вище ЛЗ. У разі супутнього швидко прогресуючого інтерстиціального захворювання легенів (особливо за наявності антитіл до MDA5) розгляньте можливість в/в введення ГК і комбіновану терапію з 2 з наступних: ритуксимаб, циклофосфамід, ВВІГ, мікофенолату мофетил, інгібітор кальциневрину, інгібітор JAK.
У процесі відновлення функціональної здатності важливою є кінезотерапія. У гострій фазі захворювання в основному слід застосовувати пасивні вправи, які запобігають виникненню контрактур; під час реконвалесценції — ізометричні вправи і вправи для розвантаження, пізніше додатково ізотонічні і аеробні вправи; дуже корисними є вправи у воді. Фізичне навантаження у цей період не повинно перевищувати 60 % максимального використання кисню.
Моніторингвгору
Періодична оцінка м'язової сили, активності КФК у сироватці, ЕМГ, а в пацієнтів з інтерстиціальним захворюванням легень — КТВРЗ, функціональні дослідження легень і тест з 6-хвилинною ходьбою. Важливо є зберігати онкологічну настороженість з огляду на підвищений ризик розвитку злоякісних пухлин. Злоякісні пухлини, асоційовані з ДМ, зустрічаються у ≈30–40 % пацієнтів, а ризик розвитку злоякісних пухлин у 6 разів вищий, ніж у загальній популяції. З цієї причини ДМ вважається паранеопластичним синдромом. Індукований новоутворенням ДМ визначається як ДМ, який розвинувся протягом року після діагностування новоутворення, або коли новоутворення було виявлено протягом 3-х років від дебюту ДМ. Частіше виникають карциноми яєчника, молочної залози, легенів, шлунка, кишківника, носової порожнини та горла, підшлункової залози, сечового міхура та неходжкінські лімфоми. Ризик розвитку новоутворення вищий у пацієнтів з антитілами до транскрипційого інтермедіального фактору 1 γ (TIF1γ) та до нуклеарного матриксного протеїну 2 (NXP2).
Ризик розвитку онкологічного захворювання у пацієнтів із ПМ вищий, ніж у загальній популяції, проте ПМ не вважається паранеопластичним синдромом.
ПРОГНОЗвгору
У разі правильного лікування 10-річна виживаність складає >80 %. Прогноз погіршують в т. ч. старший вік і ураження внутрішніх органів, особливо легень, наявність супутньої злоякісної пухлини, наявність деяких антитіл.



