 Español
Español
 English
English
 українська
українська
Siglas y abreviaturas: EPI — enfermedad pulmonar intersticial, FPI — fibrosis pulmonar idiopática, NII — neumonía intersticial idiopática, NIU — neumonía intersticial usual, TC — tomografía computarizada
Introducción
La fibrosis pulmonar idiopática (FPI) es una de las muchas enfermedades pulmonares intersticiales (EPI). Según la clasificación vigente, la FPI es una de las neumonías intersticiales idiopáticas (NII) de curso fibrosante crónico (tabla 1).
Se conocen algunas de las causas de EPI, como la exposición a factores ambientales nocivos, fármacos o las enfermedades sistémicas del tejido conectivo. Las EPI de etiología desconocida se denominan idiopáticas. Un ejemplo de este grupo es la FPI.
En la actualidad existen algunas hipótesis sobre la etiopatogenia de la FPI, pero ninguna de ellas ha sido demostrada de manera definitiva. Las pruebas que tienen como objetivo establecer las causas potenciales de la FPI se refieren a:
1) infecciones crónicas, sobre todo víricas (VEB, virus del herpes simple, VHC)1-5
2) reflejo gastroesofágico6,7
3) factores genéticos (el riesgo de fibrosis es mayor p. ej. por las mutaciones de genes del complejo de la telomerasa, de genes que codifican la proteína C y A2 surfactante y el polimorfismo del gen MUC5B).8,9 En un 5 % de los enfermos la fibrosis es hereditaria.10
El riesgo de padecer FPI aumenta con la edad.13 En el momento del diagnóstico la mayoría de los enfermos tiene >60 años, y la edad media se estima en 66 años.14,15 Con mayor frecuencia enferman hombres.14,16 Suelen ser personas con antecedentes de haber fumado o ser fumadores activos. El riesgo de FPI aumenta en personas que fuman >20 paquetes-año.16,17 La EPI es una enfermedad rara. La FPI es la NII más frecuente y la segunda EPI más frecuente después de la sarcoidosis.
Los avances en el conocimiento y desarrollo de las técnicas de imagen han influido en los criterios diagnósticos y en las recomendaciones sobre la FPI. Las últimas guías internacionales han sido publicadas en 2018.14 Más adelante se cita la información práctica más importante de este documento.
Para diagnosticar la FPI hace falta:
1) confirmar la presencia de un cuadro clínico compatible
2) confirmar la presencia de alteraciones pulmonares estructurales con patrón de NIU en TC de alta resolución, o considerando los cambios radiológicos junto a los histopatológicos
3) descartar otras causas.
Cuadro clínico
Los enfermos con FPI con frecuencia se quejan de disnea (inicialmente) de esfuerzo y/o de tos persistente (más frecuentemente seca), que se intensifican gradualmente en el curso de varios meses. Asimismo pueden aparecer síntomas inespecíficos tales como debilidad y pérdida de peso. La probabilidad de diagnosticar la FPI aumenta si se observan crepitantes basales bilaterales en la auscultación, sobre todo asociadios a acropaquia. Las alteraciones radiológicas dependen del estadio de la enfermedad. Inicialmente tienen forma de opacidades reticulares finas, que en las fases más tempranas se aprecian únicamente en la radiografía lateral en los ángulos castofrénicos posteriores. Con la progresión de la enfermedad se aprecian signos de fibrosis con imagen de panal de abejas, bronquiectasias por tracción, reducción del tamaño de los campos pulmonares y elevación de las cúpulas diafragmáticas. En las pruebas funcionales se suele observar reducción de la capacidad de difusión pulmonar del monóxido de carbono (DLCO) y a veces un patrón ventilatorio restrictivo en la espirometría y/o restricción en la pletismografía (aunque no es una alteración necesaria para diagnosticar la enfermedad).
Los síntomas de FPI no son característicos, por lo que el diagnóstico diferencial requiere excluir otras enfermedades que cursan con disnea (p. ej. enfermedad pulmonar obstructiva crónica, que también cursa con disnea de esfuerzo como síntoma principal, así como tabaquismo), o insuficiencia cardíaca (debido a los crepitantes sobre los campos inferiores). Asimismo hay que recordar que la FPI se presenta sobre todo en personas de edad avanzada, en las que con frecuencia coexisten otras enfermedades que pueden dificultar tanto el diagnóstico como la decisión de establecer tratamiento antifibrótico.
El inicio de la enfermedad puede ser insidioso y los cambios pueden progresar lentamente, por lo que por un tiempo prolongado no se notan o se subestiman. La enfermedad con mayor frecuencia tiene un curso progresivo constante. Debido al carácter irreversible de las lesiones, el pronóstico es malo. La mediana de supervivencia se estima en 3-5 años. Muy raramente la FPI tiene un inicio brusco (la denominada exacerbación súbita) o comienza a edades jóvenes, sobre todo en casos de fibrosis familiar.18 En el curso de la enfermedad pueden presentarse exacerbaciones, que suelen acelerar considerablemente la reducción de la función pulmonar, y empeoran el pronóstico.
Diagnóstico diferencial
Un elemento importante del diagnóstico de FPI es la exclusión de otras causas conocidas de alteraciones pulmonares intersticiales. Se trata de: enfermedades sistémicas del tejido conectivo, sarcoidosis, antecedentes de enfermedades neoplásicas y tratamiento con citostáticos, fármacos administrados crónicamente (también en el pasado, p. ej. amiodarona), exposición ambiental, profesional, cría de animales. No hay pruebas de laboratorio que permitan diagnosticar la FPI, pero algunas son útiles para establecer otras causas de fibrosis pulmonar intersticial (anticuerpos antinucleares, factor reumatoide, anticuerpos anticitrulina, anticuerpos contra los antígenos presentes en polvos orgánicos). Si no se consigue identificar la causa de los cambios pulmonares intersticiales, se puede pasar a siguientes etapas de diagnóstico dirigido hacia FPI.
Tras excluir otras causas, el segundo paso en el diagnóstico de la FPI es la presencia de cambios morfológicos en los pulmones con características de neumonía intersticial usual (NIU). Hay que subrayar que las alteraciones de la arquitectura pulmonar de este tipo no son patognomónicas de FPI, ya que pueden ser causadas por otras enfermedades (p. ej. sarcoidosis, alveolitis alérgica, enfermedades del tejido conectivo), pero su presencia es imprescindible para el diagnóstico. El cuadro de NIU puede confirmarse de dos maneras: mediante TC de alta resolución y realizando una biopsia pulmonar. La primera prueba que se realiza es la prueba no invasiva, es decir la TC de alta resolución. El esquema de diagnóstico recomendado en las guías de ATS/ERS/JRS/ALAT 2018 se presenta en la fig. 1.
TC de alta resolución
La TC de alta resolución permite una valoración precisa del carácter, distribución y extensión de las lesiones en el curso de una EPI. Un radiólogo experimentado puede realizar el diagnóstico (seguro, típico) de NIU, así como indicar si el caso requiere un diagnóstico diferencial.19,20 La técnica recomendada es la tomografía espiral multicorte, basada en la imagen volumétrica de las capas sobrepuestas, lo que permite p. ej. una reconstrucción multiplanar y una valoración radiológica más precisa del carácter de las lesiones. Usualmente, la prueba se realiza durante el pico inspiratorio en decúbito supino, sin embargo en la fase temprana de la fibrosis se recomienda también realizar la prueba en decúbito prono. En el diagnóstico diferencial pueden ser útiles también pruebas adicionales realizadas durante la espiración, que permiten identificar áreas de atrapamiento aéreo. Para el diagnóstico de FPI, hay que verificar si la TC de alta resolución cumple con los requerimientos técnicos recomendados en las guías. El patrón de NIU en la imagen de TC de alta resolución está compuesto por:
1) patrón reticular fino
2) distribución típica predominante a nivel subpleural y en bases
3) patrón en panal de abejas consistente en la aparición de pequeños espacios aéreos, generalmente de tamaño similar (3-10 mm), de localización subpleural, con paredes bien definidas21), con o sin presencia de bronquiectasias o dilatación de brionquiolos por tracción (fig. 2)
4) ausencia de cambios no explicados por la NIU (tales como una distribución predominante en los campos pulmonares superiores y medios, a lo largo de bronquios y vasos, extensas áreas en vidrio esmerilado, cambios micronodulares extensos, quistes múltiples bilaterales fuera de las áreas de panalización, imagen en mosaico difuso/atrapamiento aéreo, fibrosis densa en segmentos/lóbulos).
Si en la TC de alta resolución se observan signos de NIU en un enfermo en el que se han excluido otras causas posibles de cambios intersticiales, la FPI puede diagnosticarse sin otras pruebas invasivas posteriores (basta con la colaboración del médico clínico y del radiólogo). No en todos los enfermos es posible identificar todos los criterios radiológicos de NIU mencionados más arriba. Las guías actuales sugieren cuatro categorías de alteraciones que indican diferente probabilidad diagnóstica de FPI (tabla 2).
Lavado broncoalveolar
En el esquema diagnóstico actual de FPI se incluye también el lavado broncoalveolar (LBA). Sin embargo, esta técnica tiene un valor limitado, siendo más útil en el diagnóstico diferencial, sobre todo en casos de sospecha de eosinofilias pulmonares o infecciones.14
Biopsia pulmonar
Otro método para confirmar lesiones pulmonares indicativas de NIU es el estudio histopatológico. Hay que considerarla en enfermos en los que las alteraciones en la TC de alta resolución no permiten un diagnóstico “de certeza“ de NIU (p. ej. ausencia de signos de panalización). Sin embargo, es un procedimiento de alto riesgo en enfermos con sospecha de FPI. Por un lado, facilita el diagnóstico y ayuda a establecer el tratamiento antifibrótico; pero por otro lado aumenta el riesgo de exacerbaciones de la FPI y de complicaciones de la intervención (enfermos de edad avanzada, a menudo con enfermedades concomitantes). El análisis de los resultados de numerosos ensayos indica que la probabilidad de diagnosticar FPI es elevada incluso en enfermos con patrón de NIU probable en la TC de alta resolución, si está presente un contexto clínico compatible (edad >60 años, sin exposición ambiental significativa, influencia de fármacos, signos de enfermedades del tejido conectivo22). En los demás casos hay que valorar la realización de biopsia. Cambios microscópicos más importantes que permiten el diagnóstico de NIU en el estudio histopatológico:
1) focos difusos de fibrosis colagénica, separados por áreas de tejido pulmonar normal (sano)
2) áreas de destrucción completa y remodelación del tejido con pequeños espacios aéreos quísticos (panal de abejas)
3) focos fibroblásticos caracterizados por fibrosis activa, en general localizada en márgenes de fibrosis, en el límite de tejido pulmonar aéreo.
Al igual que en el caso de la TC de alta resolución, también la imagen histológica puede cumplir los criterios del patrón de NIU en diferente grado (tabla 2).
Papel del equipo multidisciplinar
Las guías actuales subrayan el papel del debate en un equipo multidisciplinar, recomendado en todos los enfermos diagnosticados de EPI de causa desconocida, con sospecha clínica de FPI.12 Se ha demostrado que tal debate aumenta la posibilidad de realizar un diagnóstico correcto de EPI que cursa con fibrosis.23-28 Tiene mayor importancia en casos sin diagnóstico radiológico de certeza de NIU (en la TC de alta resolución).
Si la imagen radiológica no cumple los criterios de NIU, la FPI puede diagnosticarse sobre la base de la combinación de los resultados de la TC de alta resolución y de la prueba histopatológica (tabla 3).
Resumen
Los principios de actuación recomendados para el diagnóstico de enfermos con sospecha de FPI se citan en la fig. 3.
1. En todo enfermo con sospecha de FPI debería indagarse detalladamente sobre las causas conocidas de cambios intersticiales pulmonares. En caso de sospecha de un origen secundario de las lesiones, el diagnóstico debe ampliarse en dicha dirección (p. ej., consultando con un reumatólogo).
2. Si la imagen de la TC de alta resolución cumple los criterios de NIU sin suscitar dudas, no se deben realizar procedimientos diagnósticos invasivos.
3. En caso de imágenes en la TC de alta resolución diferentes a la NIU, está indicado consultar a un equipo multidisciplinar, así como considerar procedimientos diagnósticos invasivos.
4. No se deben medir biomarcadores de fibrosis (recomendación fuerte), ya que no está comprobado que sea útil para el diagnóstico, ni para la indicación terapéutica.
La identificación correcta de los enfermos con FPI, así como un diagnóstico correcto, tiene gran importancia debido a la posibilidad de terapia con fármacos de acción antifibrótica (pirfenidona y nintedanib). Los enfermos con sospecha de FPI deberían dirigirse a centros especializados para un diagnóstico e inicio rápido del tratamiento. Asimismo cabe subrayar que está definitivamente contraindicado el tratamiento inmunosupresor de muy larga duración (prednisona con azatioprina y N-acetilcisteína) en enfermos con FPI, ya que aumenta el riesgo de muerte en casi 10 veces.29
A recordar
- El diagnóstico de FPI en una persona con sospecha clínica requiere confirmar la presencia de alteraciones estructurales pulmonares en forma de neumonía intersticial usual (NIU) por TC de alta resolución o una combinación adecuada de alteraciones en TC de alta resolución y cambios histopatológicos, así como excluir otras causas conocidas de dichos cambios.
- Los enfermos con sospecha de FPI deben dirigirse a centros especializados para un diagnóstico y tratamiento adecuados.
|
De causa conocida |
|
Inducida por fármacos |
|
Enfermedades del tejido conectivo |
|
Vasculitis sistémicas |
|
Factores ambientales |
|
Neumonías intersticiales idiopáticas (NII) |
|
Básicas: |
|
–fibrosis pulmonar idiopática (FPI) |
|
– neumonía intersticial idiopática inespecífica (NIII) |
|
– enfermedad pulmonar intersticial con bronquiolitis respiratoria (RB-ILD) |
|
– neumonía intersticial descamativa (DIP) |
|
– neumonía organizada criptogénica (COP) |
|
Poco frecuentes: |
|
– neumonía intersticial linfocítica (LIP) |
|
– fibroelastosis pleuroparenquimatosa idiopática (PPFE) |
|
No clasificadas |
|
En el curso de enfermedades granulomatosas |
|
Sarcoidosis |
|
Neumonía por hipersensibilidad (HP, antes alveolitis alérgica) |
|
Otras raras |
|
Linfangioleiomiomatosis (LAM) |
|
Histiocitosis de células de Langerhans |
|
Neumonía eosinofílica |
|
Proteinosis pulmonar |
|
Categorías de alteraciones |
Criterios de la TC de alta resolución |
Criterios morfopatológicos |
|
NIU |
Patrón reticular fino, de localización predominantemente subpleural y basal Panal de abejas con o sin bronquiectasia/dilatación de bronquiolos por tracción |
Fibrosis densa y endurecida con alteración de la arquitectura (es decir, cicatrización destructiva y/o panal de abejas) Cambios sobre todo subpleurales y/o a lo largo del septo interlobulillar Fibrosis focalizada Focos fibroblásticos presentes Ausencia de cambios que sugieran otro diagnóstico |
|
NIU probable |
Patrón reticular fino, de localización predominantemente subpleural y basal Presencia de bronquiectasias/dilatación de bronquiolos por tracción Posible presencia de alteraciones en vidrio esmerilado de baja intensidad |
Están presentes algunos de los rasgos histológicos enumerados más arriba, pero de forma insuficiente para establecer un diagnóstico cierto de NIU/FPI. Están ausentes cambios que indiquen otro diagnóstico, o están presenten solamente las alteraciones en panal de abeja |
|
No concluyente para la NIU |
Alteraciones predominantemente subpleurales y basales Patrón reticular fino, posible presencia de alteraciones poco intensas en vidrio esmerilado, y deformaciones bronquiales ("NIU temprana") El carácter y la distribución de los cambios no sugieren etiología alguna ("patrón verdaderamente indeterminado") |
Fibrosis con (o sin) alteraciones de la arquitectura parenquimatosa, con signos predominantes de otras alteraciones diferentes la NIU, o rasgos de NIU secundaria a otras causasa Algunas características histológicas de la NIU, pero con rasgos sugerentes de otro diagnósticob |
|
Diagnóstico alternativo |
Alteraciones o distribución característica de otras enfermedades intersticiales (quistes, nódulos, imagen en mosaico, consolidaciones, alteraciones en vidrio esmerilado, localización peribronquial, linfadenopatías, derrame pleural/alteraciones linfáticas, esofágicas, costales, en los lóbulos superiores y/o medios) |
Rasgos microscópicos que indican otra NII (p. ej. no hay focos fibroblásticos ni fibrosis) en todas las muestras Rasgos histológicos que indican otro diagnóstico (p. ej. alveolitis alérgica, histiocitosis, sarcoidosis, LAM) |
|
a Granulomas, membranas hialinas, alteraciones peribronquiolares, neumonía organizada, neumonía intersticial sin fibrosis. b Infiltración inflamatoria celular separada de las áreas en panal de abeja, hiperplasia linfoide prominente, distribución claramente bronquiolocéntrica, que puede abarcar una metaplasia peribronquiolar extensa. | ||
|
Prueba histológica TC de alta resolución |
NIU |
NIU probable |
No concluyente para la NIU |
Diagnóstico alternativo |
NIU |
FPI |
FPI |
FPI |
No FPI |
|
NIU probable |
FPI |
FPI |
FPI probable |
No FPI |
|
No concluyente para la NIU |
FPI |
FPI probable |
Fibrosis indeterminada |
No FPI |
|
Diagnóstico alternativo |
FPI posible/no FPI |
No FPI |
No FPI |
No FPI |
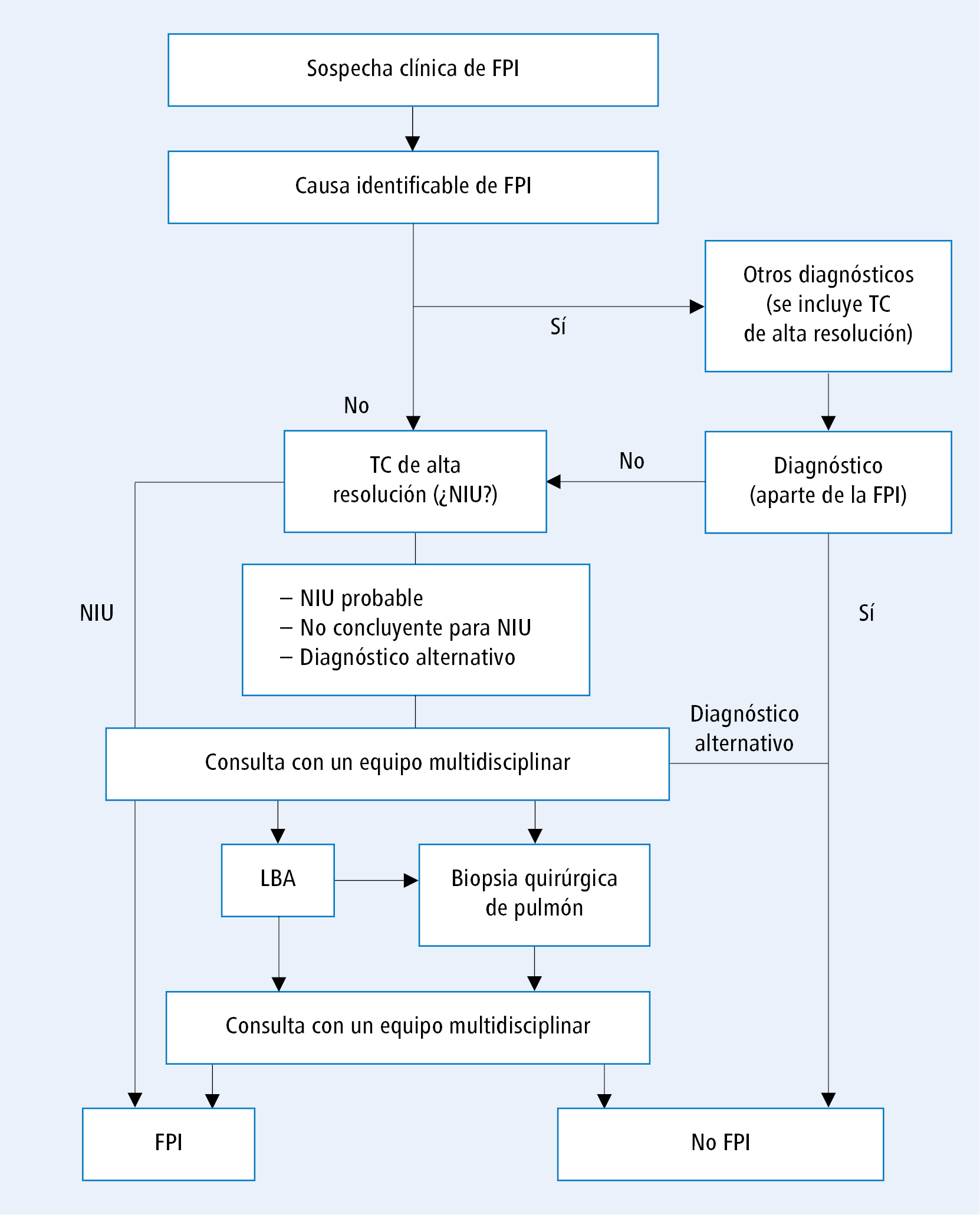
Fig. 1. Esquema diagnóstico de la FPI (según las guías de ATS/ERS/JRS/ALAT 2018, modificadas)
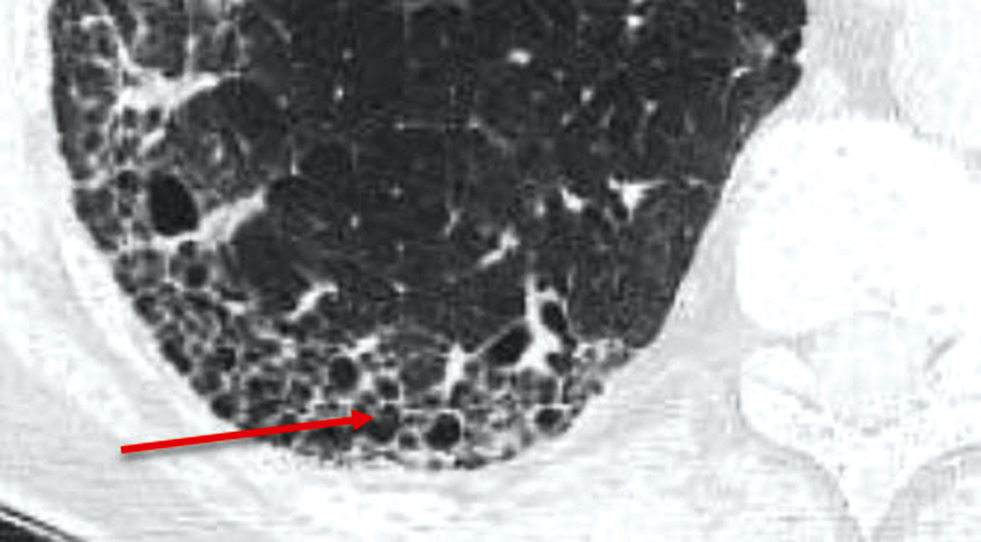
Fig. 2. Imagen de panal de abejas en la TC de alta resolución
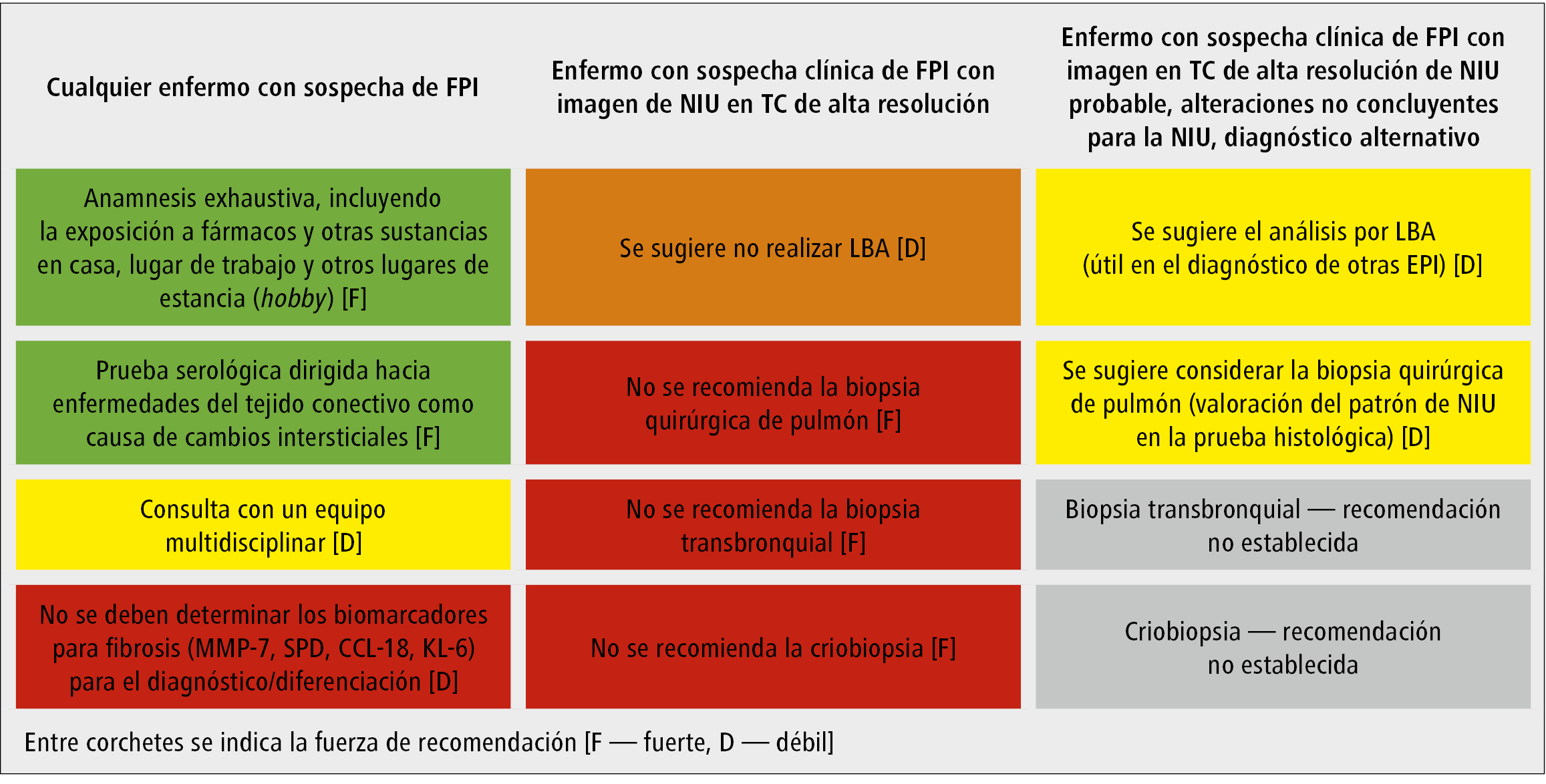
Fig. 3. Recomendaciones para las pruebas utilizadas en el diagnóstico de FPI (según las guías de ATS/ERS/JRS/ALAT14)
Bibliografía:
1. Ueda T., Ohta K., Suzuki N. y cols., Idiopathic pulmonary fibrosis and high prevalence of serum antibodies to hepatitis C virus, Am. Rev. Respir. Dis., 1992; 146: 266-268.2. Egan J.J., Stewart J.P., Hasleton P.S. y cols., Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis, Thorax, 1995; 50: 1234-1239.
3. Wangoo A., Shaw R.J., Diss T.C. y cols., Cryptogenic fibrosing alveolitis: lack of association with Epstein-Barr virus infection, Thorax, 1997; 52: 888-891.
4. Tang Y.W., Johnson J.E., Browning P.J. y cols., Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis, J. Clin. Microbiol., 2003; 41: 2633-2640.
5. Lasithiotaki I., Antoniou K.M., Vlahava V.M. y cols., Detection of herpes simplex virus type1 in patients with fibrotic lung diseases, PLoS One, 2011; 6: e27 800.
6. Lee J.S., Collard H.R., Raghu G. y cols., Does chronic microaspiration cause idiopathic pulmonary fibrosis?, Am. J. Med., 2010; 123: 304-311.
7. Downing L., Sawarynski K.E., Li J. y cols., A simple quantitative method for assessing pulmonary damage after x irradiation, Radiat. Res., 2010; 173: 536-544.
8. Talbert J.L., Schwartz D.A., Steele M.P., Familial Interstitial Pneumonia (FIP), Clin. Pulm. Med., 2014; 21: 120-127.
9. Seibold M.A., Wise A.L., Speer M.C. y cols., A common MUC5B promoter polymorphism and pulmonary fibrosis, N. Engl. J.Med., 2011; 364: 1503-1512.
10. Raghu G., Collard H.R., Egan J.J. y cols., An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidencebased guidelines for diagnosis and management, Am. J. Respir. Crit. Care Med., 2011; 183: 788-824.
11. American Thoracic Society, European Respiratory Society: American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001, Am. J. Respir. Crit. Care Med., 2002; 165: 277-304.
12. Travis W.D., Costabel U., Hansell D.M. y cols., An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias, Am. J. Respir. Crit. Care Med., 2013; 188: 733-748.
13. Le J., Gribbin J., West J. y cols., The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK, Respir. Med., 2007; 101: 2534-2540.
14. Raghu G., RemyJardin M., Myers J.L. y cols., Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline, Am. J. Respir. Crit. Care Med., 2018; 198: e44-e68.
15. Salisbury M.L., Xia M., Murray S. y cols., Predictors of idiopathic pulmonary fibrosis in absence of radiologic honeycombing: a cross sectional analysis in ILD patients undergoing lung tissue sampling, Respir. Med., 2016; 118: 88-95.
16. Behr J., Kreuter M., Hoeper M.M. y cols., Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTSIPF registry, Eur. Respir. J., 2015; 46: 186-196.
17. Baumgartner K.B., Samet J.M., Stidley C.A. y cols., Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med., 1997; 155: 242-248.
18. Allen R.J., Porte J., Braybrooke R. y cols., Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genomewide association study, Lancet Respir. Med., 2017; 5: 869-880.
19. Wolter N.J., Kunkel S.L., Lynch J.P. III, Ward P.A., Production of cyclooxygenase products by alveolar macrophages in pulmonary sarcoidosis, Chest, 1983; 83 (5 suppl.): 79S-81S.
20. Johkoh T., Muller N.L., Cartier Y. y cols., Idiopathic interstitial pneumonias: diagnostic accuracy of thin section CT in 129 patients, Radiology, 1999; 211: 555-560.
21. Hansell D.M., Bankier A.A., MacMahon H. Fleischner Society: glossary of terms for thoracic imaging, Radiology, 2008; 246: 697-722.
22. Lynch D.A., Sverzellati N., Travis W.D. y cols., Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper, Lancet Respir. Med., 2018; 6: 138-153.
23. Flaherty K.R., King T.E. Jr, Raghu G. y cols., Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis?, Am. J. Respir. Crit. Care Med., 2004; 170: 904-910.
24. Flaherty K.R., King T.E. Jr y cols., Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis?, Am. J. Respir. Crit. Care Med., 2007; 175: 1054-1060.
25. Chaudhuri N., Spencer L., Greaves M. y cols., A review of the multidisciplinary diagnosis of interstitial lung diseases: a retrospective analysis in a single UK specialist centre, J. Clin. Med., 2016; 5.
26. Thomeer M., Demedts M., Behr J. y cols., Multidisciplinary interobserver agreement in the diagnosis of idiopathic pulmonary fibrosis, Eur. Respir. J., 2008; 31: 585-591.
27. Theegarten D., Muller H.M., Bonella F. y cols., Diagnostic approach to interstitial pneumonias in a single centre: report on 88 cases, Diagn. Pathol., 2012; 7: 160.
28. Jo H.E., Glaspole I.N., Levin K.C. y cols., Clinical impact of the interstitial lung disease multidisciplinary service, Respirology, 2016; 21: 1438-1444.
29. Idiopathic Pulmonary Fibrosis Clinical Research: Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis, N. Engl. J. Med., 2012; 366: 1968-1977.

