 English
English
 Español
Español
 українська
українська

Tiede A, Collins P, Knoebl P, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. 2020 Jul;105(7):1791-1801. doi: 10.3324/haematol.2019.230771. Epub 2020 May 7. PMID: 32381574; PMCID: PMC7327664.
Holstein K, Liu X, Smith A, Knöbl P, et al. Bleeding and response to hemostatic therapy in acquired hemophilia A: results from the GTH-AH 01/2010 study. Blood. 2020 Jul 16;136(3):279-287. doi: 10.1182/blood.2019003639. PMID: 32268359; PMCID: PMC7392842.
Definition, Etiology, PathogenesisTop
Acquired hemophilia A (AHA) is an autoimmune disease caused by the development of neutralizing antibodies to factor VIII (factor VIII inhibitors). AHA is very rare and affects both men and women. Etiology is unknown in 50% of patients. AHA may develop during pregnancy or within 12 months after delivery as well as in patients with autoimmune diseases, malignancy, allergic diseases, or drug-induced reactions. It has been observed that there are 2 peaks of AHA incidence: one in young women, associated with pregnancy and the second one at advanced age (>60 years).
Clinical Features and Natural HistoryTop
AHA manifests as acute severe coagulopathy with cutaneous, mucous membrane (gastrointestinal, urinary, genital), dental site, and surgical site bleeding in a patient without a previous history of bleeding. Less common features include retroperitoneal hematoma, intracranial bleeding, and bleeding into skeletal muscles. Very rarely, patients may have spontaneous bleeding into the joints (a manifestation typical for congenital hemophilia A).
In ~30% of patients (and more frequently for postpartum cases) AHA resolves spontaneously within one year. Relapses occur in ~20% of patients in whom remission has been achieved with prior immunosuppressive treatment.
DiagnosisTop
1. Screening tests: Activated partial thromboplastin time (aPTT) is typically prolonged. Prothrombin time (PT), thrombin time (TT), platelet counts, and serum fibrinogen levels are normal (unless consumption or dilutional coagulopathy has developed or the inhibitor antibody affects multiple coagulation factors).
2. Studies confirming the diagnosis: Absence of aPTT normalization in a 1:1 mixture of the patient’s plasma and normal plasma (the aPTT mixing test, which requires prolonged incubation to detect acquired inhibitors), decreased factor VIII activity (usually 2-10 IU/dL), a positive factor VIII inhibitor test result. Confirmation of the antibody should be performed in a laboratory with specific expertise, given the variability in the activity of the antibodies that cause this disorder.
AHA is diagnosed on the basis of typical clinical manifestations as well as results of the screening tests and studies confirming the diagnosis (Figure 9.2-1).
A combination of screening test results consistent with AHA may also be seen in hemophilia A, inherited deficiency of factor XI or factor XII, or in patients with lupus anticoagulant (in which case thrombosis may be present). Various acquired coagulopathies can present with similar signs and symptoms (eg, well-compensated chronic disseminated intravascular coagulation).
TreatmentTop
Management of a patient with AHA: Figure 9.2-2.
1. Short-term treatment goals include treatment of bleeding.
2. The long-term treatment goal is eradication of factor VIII inhibitors.
1. Promptly diagnose and treat underlying conditions, if any.
2. Avoid invasive procedures, IM injections, administration of acetylsalicylic acid (ASA) or nonsteroidal anti-inflammatory drugs (NSAIDs). Be conservative in performing fasciotomy for compartmental syndromes provoked by muscular hematomas.
1. Treatment of bleeding (all patients should be treated at an expert center or in consultation with experts in this area): Recombinant activated factor VII concentrate (rFVIIa) ≥90 microg/kg IV every 2 to 24 hours, activated prothrombin complex concentrate (aPCC) 50 to 100 IU/kg IV every 8 to 12 hours (up to a maximum dose of 200 IU/kg per day), or recombinant porcine factor VIII 50 to 200 IU/kg per day.
2. Immunosuppressive treatment should be started immediately after the diagnosis of AHA is made. Consider possible contraindications and the risk of adverse events, including severe infections:
1) The first-line treatment is oral prednisone 1 mg/kg/d for 3 to 4 weeks (max. 4-6 weeks); it may be combined with rituximab, oral cyclophosphamide 1.5 to 2 mg/kg/d, or mycophenolate mofetil in patients not responding to the treatment with steroids. Remission is achieved in ~70% of patients, for whom very slow tapering of prednisone is recommended.
2) In patients not responding to immunosuppressive treatment, continue the follow-up and treat bleeding when present.
3) In patients with relapse use the immunosuppressive treatment that achieved the first remission.
FiguresTop
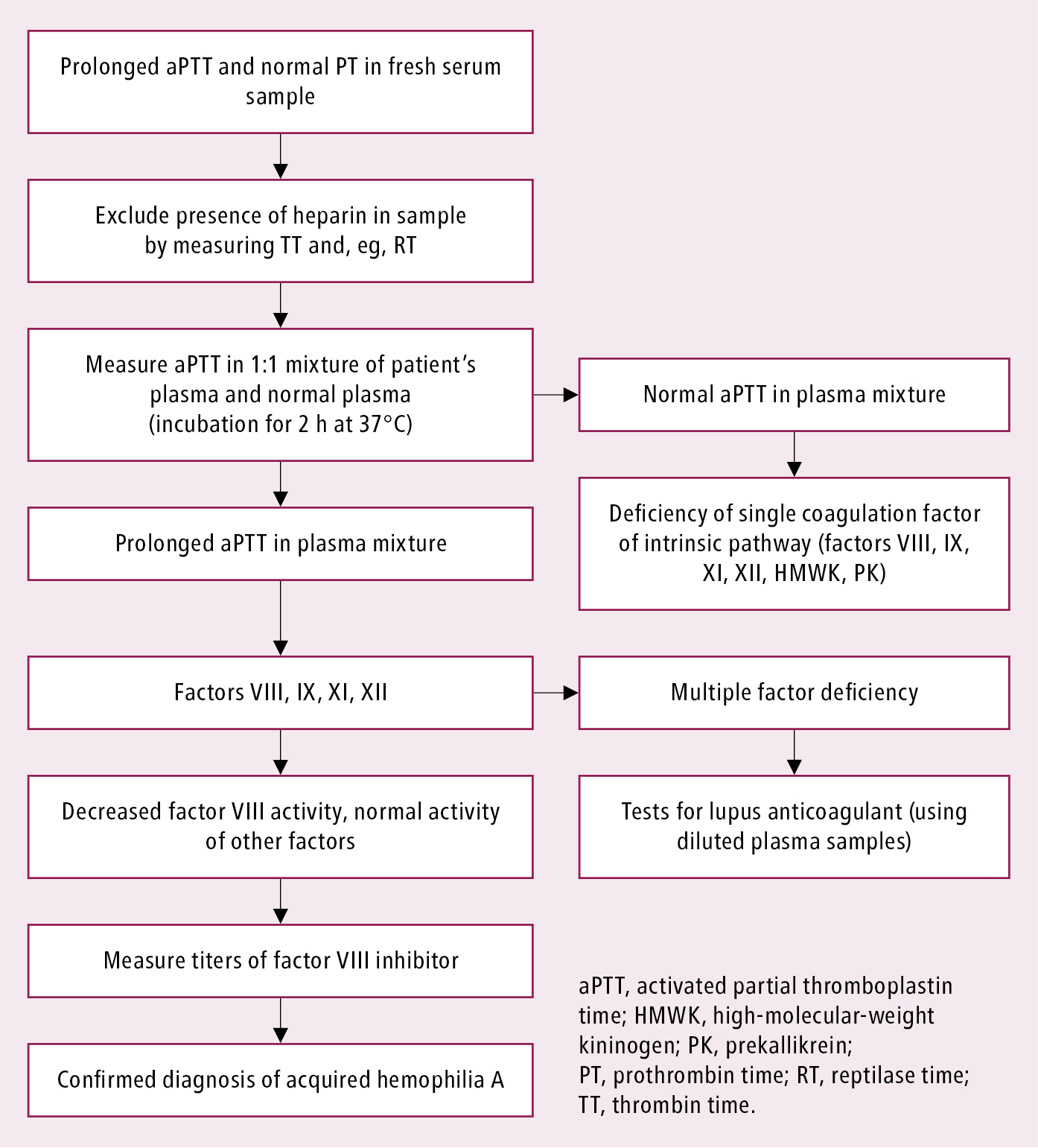
Figure 9.2-1. Diagnostic algorithm of acquired hemophilia.
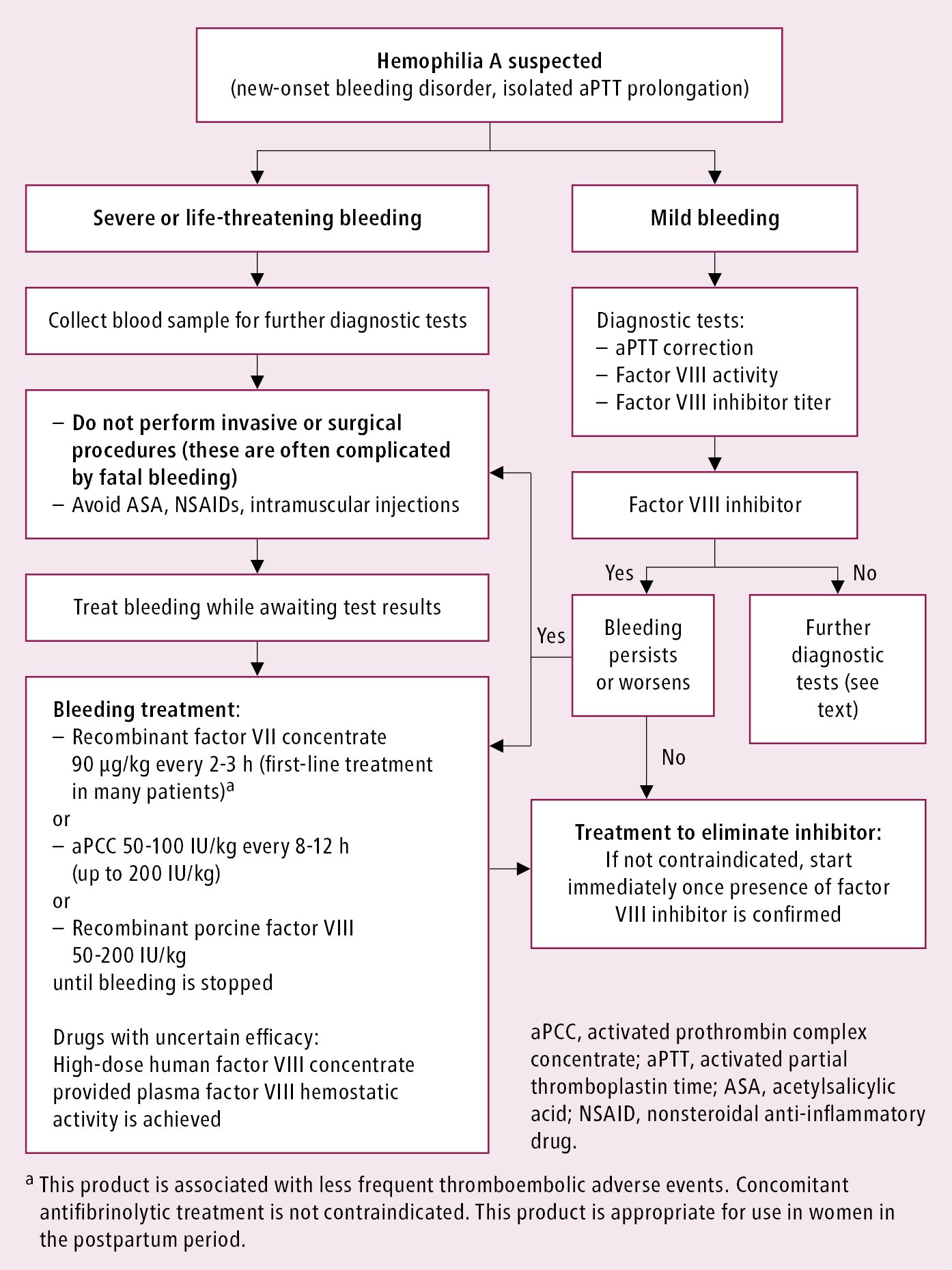
Figure 9.2-2. Management of a patient with acquired hemophilia A.