 English
English
 Español
Español
 українська
українська

Goldust M, Hagstrom EL, Rathod D, Ortega-Loayza AG. Diagnosis and novel clinical treatment strategies for pyoderma gangrenosum. Expert Rev Clin Pharmacol. 2020 Feb;13(2):157-161. doi: 10.1080/17512433.2020.1709825. PMID: 31875484.
Borda LJ, Wong LL, Marzano AV, Ortega-Loayza AG. Extracutaneous involvement of pyoderma gangrenosum. Arch Dermatol Res. 2019 Aug;311(6):425-434. doi: 10.1007/s00403-019-01912-1. PMID: 30923901.
Ben Abdallah H, Fogh K, Bech R. Pyoderma gangrenosum and tumour necrosis factor alpha inhibitors: A semi-systematic review. Int Wound J. 2019 Apr;16(2):511-521. doi: 10.1111/iwj.13067. Epub 2019 Jan 3. PMID: 30604927.
Shavit E, Alavi A, Sibbald RG. Pyoderma Gangrenosum: A Critical Appraisal. Adv Skin Wound Care. 2017 Dec;30(12):534-542. doi: 10.1097/01.ASW.0000526605.34372.9e. PMID: 29140836.
Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma Gangrenosum: An Update on Pathophysiology, Diagnosis and Treatment. Am J Clin Dermatol. 2017 Jun;18(3):355-372. doi: 10.1007/s40257-017-0251-7. PMID: 28224502.
Brooklyn T, Dunnill G, Probert C. Diagnosis and treatment of pyoderma gangrenosum. BMJ. 2006 Jul 22;333(7560):181-184. doi: 10.1136/bmj.333.7560.181. PMID: 16858047; PMCID: PMC1513476.
Definition, Etiology, PathogenesisTop
Pyoderma gangrenosum (PG) is a neutrophilic ulcerative dermatosis with a spectrum of clinical presentations and variable clinical course. PG is caused by genetic alterations of the immune system (both innate and adaptive), leading to inflammasome activation, cytokine production, and neutrophilic infiltration. Genetic mutations, neutrophil dysfunction, and abnormal inflammation contribute to the pathogenesis and clinical manifestations of PG.
Clinical Features and Natural HistoryTop
PG is a heterogeneous disease with variable clinical presentations and clinical courses, which makes the diagnosis challenging. PG commonly affects women aged 20 to 50 years and occurs on the lower extremities, but it can involve any area of the body, including the head and neck in children and the genital and perianal areas in infants. Lesions are usually limited to 1 to 3, with <5% of body surface area (BSA) involved. The morphologic variants of PG include classic ulcerative (including peristomal and postsurgical), pustular, bullous, and vegetative (superficial granulomatous) PG. The course of PG can be indolent, aggressive, or fulminant. PG often demonstrates a chronic, relapsing course. Worsening or development of new lesions after a trauma (known as pathergy) is a common characteristic of active PG.
1. Classic ulcerative PG: It is the most common PG variant that most frequently affects the legs. It is characterized by a rapidly progressing painful wound with a peripheral red halo and raised red-purple undermined edges (Figure 4.6-1). The center consists of necrotic tissue with a purulent or granulomatous base with active borders. The healing stage consists of wound edges with the epithelium projecting into the ulcer, known as Gulliver sign, which heals as cigarette paper–like or cribriform scars. Patients with PG are prone to an exaggerated skin injury presenting as pathergy in 25% to 50% of cases; thus, surgery and debridement are often contraindicated. Systemic symptoms are common in classic ulcerative PG, including fever, malaise, arthralgia, and myalgia. The lung is the most common extracutaneous organ involved in patients with PG. Pulmonary involvement may range from asymptomatic presentation to severe respiratory distress.
Both peristomal and postsurgical PG are variants of the ulcerative form:
1) Peristomal PG is a form of pathergy that constitutes 15% of all PG cases. Peristomal PG ulcers occur near abdominal stomas (Figure 4.6-2), especially in those with inflammatory bowel disease (IBD), but they can also occur in those who have had urostomies, ileostomies, or colostomies for other reasons such as malignancy or diverticular disease. Peristomal PG ulcers can interfere with proper adherence of the stoma appliances to the abdominal wall; poor location or shape of the stoma are common causes for leakage and subsequent skin irritation. Treatment includes skin protection, topical steroids, systemic immunosuppressive therapy in severe cases, and relocation of the stoma when inflammation is under control.
2) Postsurgical PG occurs following any surgical trauma, with small foci of dehiscence that coalesce into an ulcer that enlarges. Postsurgical PG is a challenge for surgeons and is often misdiagnosed as postsurgical infection. The average time between surgery and onset of symptoms of postsurgical PG is 11 days; the risk of postsurgical recurrence in future surgeries of a person with PG is 15%.
2. Pustular PG is a rare, superficial PG variant beginning as a sterile inflammatory pustule or a group of sterile pustules that coalesce and ulcerate to the pustular stage with a red halo. Pustular PG ulcers are typically confined to patients with IBD and affect the trunk and extensor surfaces. These ulcers are painful and can regress without scarring or persist for months and evolve into classic PG.
3. Bullous PG is a rare PG variant that predominantly occurs on the upper limbs, face, dorsum of the hands, or extensor surfaces of the arms. Bullous PG ulcers begin concentrically, spread rapidly, and may break down to form more superficial ulcers with blue undermined edges. Bullous PG has a poor prognosis due to its association with hematologic conditions and malignancies such as acute myelogenous leukemia.
4. Vegetative (superficial granulomatous) PG is the least common PG variant. It is superficial, benign, and more responsive to local treatment compared with other PG variants. Vegetative (superficial granulomatous) PG ulcers typically present as isolated, erythematous, warty eroded and ulcerated plaques without the erythematous borders seen in classic PG. Microscopic examination of a skin biopsy specimen reveals less neutrophil infiltration and more granulomatous histology. Vegetative (superficial granulomatous) PG ulcers occur as single lesions in those who are otherwise well and typically are not associated with systemic symptoms.
5. Drug-induced PG is rare and occurs as an adverse reaction to drugs such as cocaine, levamisole (definite adverse drug reactions), isotretinoin, propylthiouracil (probably adverse drug reactions), gefitinib, imatinib, sunitinib, granulocyte colony–stimulating factor, and biologic agents. Drug-induced PG ulcers begin as painful nodules or pustules that develop into ulcers over days to weeks. Similarly as in classic PG ulcers, healing leads to cribriform scarring. Pathergy occurs in 20% to 30% of cases, especially when lesions develop at the site of drug injection. Drug-induced PG ulcers typically resolve after drug discontinuation.
6. Syndromic PG (PG occurring in discernible combination with other conditions) includes a variety of autoinflammatory syndromes such as:
1) PAPA: pyogenic arthritis, pyoderma gangrenosum, acne syndrome.
2) PASH: pyoderma gangrenosum, acne, suppurative hidradenitis syndrome.
3) PAPASH: pyogenic arthritis, pyoderma gangrenosum, acne, suppurative hidradenitis syndrome.
4) PASS: pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, seropositive spondyloarthropathies syndrome.
5) PsAPASH: psoriatic arthritis, pyoderma gangrenosum, acne, suppurative hidradenitis syndrome.
6) PAC: pyoderma gangrenosum, acne, and ulcerative colitis syndrome.
These PG-associated genetic syndromes share proinflammatory pathways and/or molecules (eg, interleukin 1 [IL-1]) underlying the pathogenesis of PG and associated conditions, such as IBD and psoriasis.
Underlying diseases such as IBD, rheumatoid arthritis, and hematologic disorders share similar proinflammatory pathogeneses with PG and are seen in up to 75% of PG cases.
Extracutaneous sites such as the lungs, eyes, kidneys, bones, spleen, and liver have been involved in PG.
1. Pulmonary involvement is the most common extracutaneous manifestation of PG and can present asymptomatically or as severe respiratory distress. Aseptic pulmonary nodules with or without cavitation are common, and interstitial lung disease with interstitial infiltrates, consolidation, mediastinal lymphadenopathy, or pleural effusions may be seen. Pulmonary involvement in PG can be fatal.
2. Ocular involvement is rare in PG. It presents as eyelid swelling with nodules early on in the disease and can progress to scleritis, proptosis, and conjunctival injection. This can further progress to purulent discharge and tissue necrosis (necrotic ulceration and fistula formation in the eyelids), which has devastating consequences including blindness.
3. Renal involvement is rare in PG and may present as pyuria, hematuria, aseptic leukocyturia, oliguria, and proteinuria, which may progress to chronic sclerosing glomerulonephritis, chronic kidney disease, end-stage renal disease, and renal carcinoma. The most common locations for PG ulcers in patients with PG and underlying chronic kidney disease and end-stage renal disease include the lower extremities, skin overlying arteriovenous fistulas, and previous surgery sites.
4. Bone involvement is rare in PG and initially presents as arthralgia, myalgia, and fever, which may progress to sterile osteomyelitis (most common), osteitis, and osteolysis.
5. Splenic involvement is exceedingly rare in PG but typically occurs with liver involvement and presents as abscesses detected on imaging.
DiagnosisTop
Due to the lack of definitive laboratory and histopathologic diagnostic criteria, PG is often misdiagnosed and is a diagnosis of exclusion. Diagnosis is typically based on clinical presentation (disease progression, nonresponsiveness to antibiotics, responsiveness to immunosuppression), as PG does not have a specific laboratory workup.
The Delphi criteria were proposed by a consensus of clinical experts based on the clinical and histopathologic findings of PG and are divided into major and minor criteria. Meeting 1 major criterion (biopsy of the ulcer edge demonstrating a neutrophilic infiltrate) and 4 of the 8 minor criteria is required for diagnosis of PG. The minor criteria include exclusion of infection on histology; positive pathergy phenomenon; personal history of IBD or inflammatory arthritis; history of a papule, pustule, or vesicle that ulcerates rapidly; presence of peripheral erythema around the ulcer; undermining border; tenderness at the site of ulceration; presence of multiple ulcerations (≥1 occurring on the anterior lower leg); cribriform scars at healed ulcer sites; and a decrease in ulcer size after immunosuppressive treatment.
1. Skin biopsy of the active border and subcutaneous tissue of the ulcer is required to exclude other diagnoses, such as primary vasculitis, inflammatory conditions, infections, and malignancy, but does not always show specific diagnostic features of PG. The heavy presence of neutrophils supports the diagnosis of PG, but this is not always observed in chronic or partially treated PG. Thus, in conjunction with a clinical presentation of PG, a nonspecific mixed inflammatory infiltrate may suggest a diagnosis of PG.
2. Tissue culture should be considered to exclude bacterial, atypical mycobacterial, and deep fungal infections that mimic PG, but it does not identify the underlying cause of PG ulcer.
The differential diagnosis of PG is broad, as there are no serologic markers or histopathologic findings specific to PG. Excluding differential diagnoses is essential in the diagnostic workup for PG. The differential diagnoses include infectious causes such as blastomycosis, sporotrichosis, leishmaniasis, ecthyma, herpes simplex virus infection, atypical mycobacterial infections, and parasitic infections; vasculitis and autoimmune causes such as Behçet disease, polyarteritis nodosa, leukocytoclastic vasculitis, cryoglobulinemia, antiphospholipid syndrome–associated ulcers, and lupus; vascular causes such as venous, arterial, and Martorell ulcers; and exogenous causes such as insect and spider bites and factitious ulcers.
TreatmentTop
PG treatment is aimed at reducing the inflammation that causes ulceration, promoting wound healing, and addressing underlying disorders. Treatment choice depends on the location, number, and size of lesions, underlying disease, cost, adverse effects, comorbidities, and patient preferences. Management includes avoidance of triggers, appropriate wound care, pain management, treatment of underlying comorbid conditions, and topical, systemic, and targeted immunomodulating therapies. Systemic glucocorticoids are the mainstay of treatment, and treating any underlying disease can help prevent rebound flares. Patients typically improve shortly after treatment initiation; however, complete resolution of the ulcer takes several months.
Wound care is essential in PG management. This includes appropriate dressing, gentle cleansing with antibacterial agents, compression to control for limb wound edema (only upon exclusion of arterial insufficiency), and maintaining a moist wound environment. Debridement (enzymatic, autolytic, or blunt surgical) must be used cautiously when removing nonviable tissues and reducing bioburden and odor. Pathergy may occur with aggressive surgical debridement and the use of strong adhesives.
1. Pain management includes topical agents, acetaminophen (INN paracetamol), nonsteroidal anti-inflammatory drugs, and opioids. Wound healing may be slowed or inhibited and the patient’s quality of life can be severely impaired without adequate pain management.
2. Local treatments are appropriate options for mild, localized, and small (<2 cm2) lesions:
1) Topical or intralesional glucocorticoids are considered first-line local therapy and can be applied to the active border of the ulcer.
2) Local calcineurin inhibitors such as topical tacrolimus are considered first-line therapy and have been used to treat PG in several case series.
3. Systemic treatments are considered first-line therapy for extensive, progressive, severe, or disfiguring disease. Patients with PG must be managed by dermatologists or rheumatologists.
1) Oral prednisone (0.5-1 mg/kg/d) is the first-line therapy for PG; treatment response is observed within 2 to 3 days. Patients should be monitored for adverse effects of long-term systemic glucocorticoid use (eg, osteopenia, weight gain, glaucoma, cataracts, hyperglycemia, diabetes, adrenal insufficiency, glucocorticoid-induced psychosis).
2) Oral cyclosporine (2.5-5 mg/kg/d) is considered second-line therapy and may be used in glucocorticoid-resistant cases or in combination with glucocorticoids. As in the case of glucocorticoid treatment, patients should be monitored for adverse effects such as renal insufficiency and hypertension.
3) IV pulsed methylprednisolone (1000 mg/d) may be used in severe and recalcitrant disease.
4) High-dose IV immunoglobulin (IVIG) (2 g/kg) over 2 to 3 consecutive days per month for ≥6 months may be used as an adjuvant treatment in recalcitrant disease, for solitary PG lesions, or when trying to prevent repetitive superinfections. Common adverse events include headache and nausea.
4. Targeted therapies include biologic agents that antagonize proinflammatory mediators in PG, such as tumor necrosis factor (TNF), IL-1 beta, and IL-6 antagonists; however, most biologic agents have been used off label for PG, with only limited and anecdotal evidence.
1) TNF antagonists:
a) Adalimumab is a potential treatment in therapy-resistant PG that has shown some clinical improvement or incomplete resolution with a good safety profile; more clinical evidence is required, as investigations are limited to small case series.
b) Infliximab has been investigated in a clinical trial on the management of PG. It is a chimeric monoclonal antibody against TNF-alpha, which inhibits cytokine production from anergic regulatory T cells.
c) Etanercept has been used in refractory PG in those with PG and active Crohn disease, but it is less efficacious than infliximab.
d) Certolizumab pegol is the newest TNF-alpha antagonist used to treat PG, leading to complete remission.
e) Golimumab is structurally similar to adalimumab, but the results of PG treatment with golimumab have been equivocal.
2) IL-1 antagonists:
a) Anakinra is a recombinant, nonglycosylated IL-1 receptor antagonist, typically used in rheumatoid arthritis and cryopyrinopathies, that has shown substantial improvement or complete resolution of PG.
b) Canakinumab is a human anti–IL-1 beta monoclonal antibody typically used in cryopyrin-associated periodic syndromes (CAPSs) and autoinflammatory syndromes associated with increased activity of caspase 1. Canakinumab has been shown to reduce wound size and improve quality of life in patients with glucocorticoid-refractory PG.
3) IL-6 antagonists: Tocilizumab has been shown to improve PG in a patient with coexisting rheumatoid arthritis.
4) IL-1 alpha antagonists: Bermekimab (RA-18C3) is the newest human IL-1 alpha antagonist in phase II investigations for the treatment of PG.
5) IL-12/23 antagonists: Ustekinumab is an IL-12/23 inhibitor shown to improve PG in several case reports.
6) Integrin inhibitors: Vedolizumab has been shown to improve PG in a patient with coexisting ulcerative colitis.
7) Cluster of differentiation 3 (CD3) inhibitors: Visilizumab has been shown to improve PG in a patient with coexisting ulcerative colitis.
8) Small molecules:
a) Tofacitinib is an inhibitor of the Janus kinase–signal transducer and activator of transcription (JAK/STAT), which has been used to treat rheumatoid arthritis in patients with PG and Crohn disease. PG resolved in these patients in whom several biologic therapies failed previously.
b) Ruxolitinib is a JAK/STAT inhibitor shown to resolve PG lesions in those with polycythemia vera and PG.
9) Phosphodiesterase-4 (PDE4) inhibitors: Apremilast has been used in a single case of recalcitrant vegetative PG. When it was used in conjunction with oral prednisone, the patient experienced complete and partial healing of 2 PG ulcers.
Management of Extracutaneous Manifestations
1. Pulmonary manifestations: If treated promptly with systemic glucocorticoids or other immunosuppressants, prognosis of pulmonary involvement in PG is favorable, with no reports of relapse.
2. Ocular manifestations: Systemic glucocorticoids are the mainstay of treatment for ocular PG.
3. Renal manifestations: Improving kidney function in conjunction with topical or intralesional and systemic glucocorticoid therapy should improve the PG ulcers.
4. Bone manifestations: Systemic glucocorticoids are the treatment of choice for both cutaneous and skeletal manifestations.
5. Splenic manifestations: Systemic immunotherapy (systemic glucocorticoids with or without systemic cyclosporine) may be used to treat splenic involvement.
PrognosisTop
Factors associated with poor prognosis include increased disease severity; older age at diagnosis; ulcerative and bullous variants; and underlying disease, superinfection, and sepsis unresponsive to treatment. Ulcer recurrence occurs in 70% of patients treated with prednisolone and in 66% of those treated with cyclosporine. Managing underlying disorders is essential in preventing rebound flares. Ultimately, despite advances in PG treatment, the prognosis is unpredictable due to the chronic, relapsing nature of PG and its complications with a mortality rate of up to 30%.
Figures Top
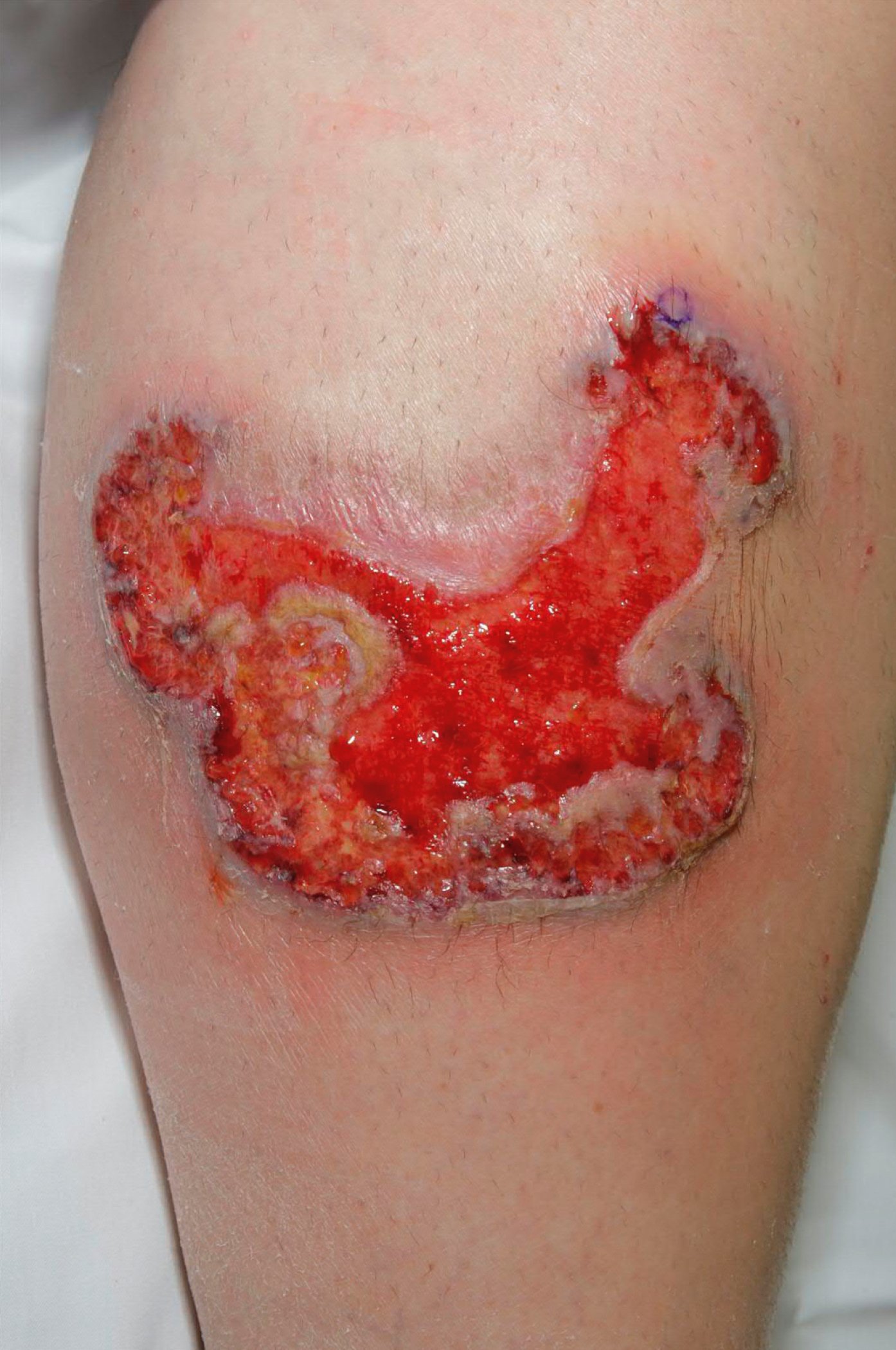
Figure 4.6-1. Ulcerative pyoderma gangrenosum. Photograph courtesy of Mayo Clinic.
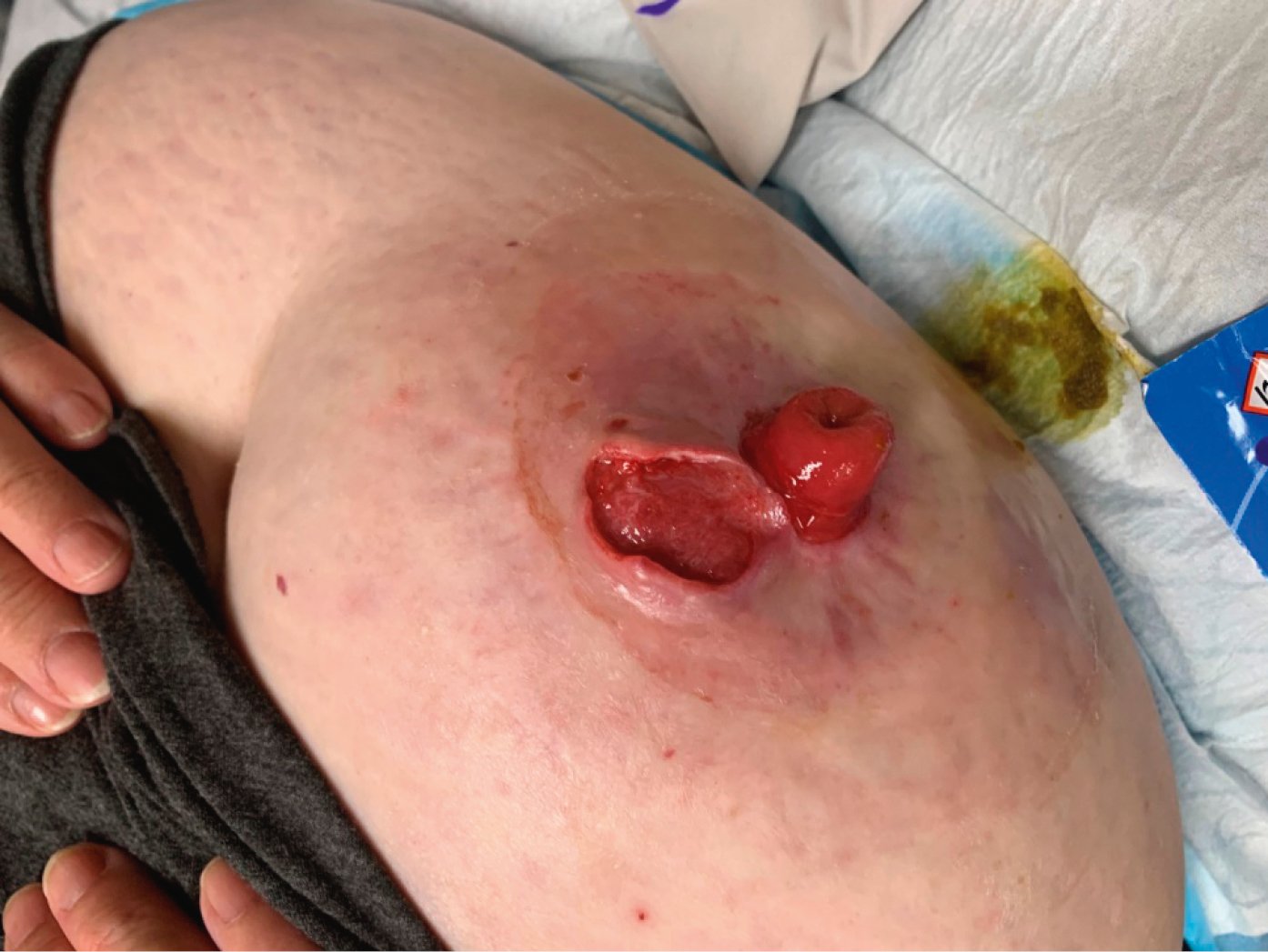
Figure 4.6-2. Peristomal pyoderma gangrenosum in a patient with Crohn disease. Photograph courtesy of Dr Mohannad Abu-Hilal.