 English
English
 Español
Español
 українська
українська

Spencer CH, Rouster-Stevens K, Gewanter H, et al; Pediatric Rheumatologist Collaborators. Biologic therapies for refractory juvenile dermatomyositis: five years of experience of the Childhood Arthritis and Rheumatology Research Alliance in North America. Pediatr Rheumatol Online J. 2017 Jun 13;15(1):50. doi: 10.1186/s12969-017-0174-0. PubMed PMID: 28610606; PubMed Central PMCID: PMC5470177.
Milone M. Diagnosis and Management of Immune-Mediated Myopathies. Mayo Clin Proc. 2017 May;92(5):826-837. doi: 10.1016/j.mayocp.2016.12.025. Review. PubMed PMID: 28473041.
Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017 Feb;76(2):329-340. doi: 10.1136/annrheumdis-2016-209247. Epub 2016 Aug 11. Review. PubMed PMID: 27515057; PubMed Central PMCID: PMC5284351.
Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EK. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin Rev Allergy Immunol. 2017 Feb;52(1):1-19. doi: 10.1007/s12016-015-8510-y. Review. PubMed PMID: 26424665; PubMed Central PMCID: PMC5828023.
Munters LA, Loell I, Ossipova E, et al. Endurance Exercise Improves Molecular Pathways of Aerobic Metabolism in Patients With Myositis. Arthritis Rheumatol. 2016 Jul;68(7):1738-50. doi: 10.1002/art.39624. PubMed PMID: 26867141.
Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic Inflammatory Myopathies: Clinical Approach and Management. Front Neurol. 2016 May 20;7:64. doi: 10.3389/fneur.2016.00064. eCollection 2016. Review. PubMed PMID: 27242652; PubMed Central PMCID: PMC4873503.
Ruperto N, Pistorio A, Oliveira S, et al; Paediatric Rheumatology International Trials Organisation (PRINTO). Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet. 2016 Feb 13;387(10019):671-8. doi: 10.1016/S0140-6736(15)01021-1. Epub 2015 Nov 30. PubMed PMID: 26645190.
Johnson NE, Arnold WD, Hebert D, et al. Disease course and therapeutic approach in dermatomyositis: A four-center retrospective study of 100 patients. Neuromuscul Disord. 2015 Aug;25(8):625-31. doi: 10.1016/j.nmd.2015.04.013. Epub 2015 May 7. PubMed PMID: 26022999; PubMed Central PMCID: PMC4519354.
Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015 Feb;42(2):282-91. doi: 10.3899/jrheum.140566. Epub 2014 Dec 1. PubMed PMID: 25448790.
Definition, Etiology, PathogenesisTop
Polymyositis (PM) is an acquired, idiopathic, inflammatory myopathy. Dermatomyositis (DM) is a type of inflammatory myopathy associated with cutaneous lesions. Both conditions may also cause inflammatory lesions of the heart, pulmonary interstitium, and in the case of DM, blood vessels.
The etiology of PM and DM is unknown; however, autoimmune and, in some cases, paraneoplastic, processes are thought to play a major role in their pathogenesis, given the frequent presence of autoantibodies and coexistent malignancies in some patients.
Clinical Features and Natural HistoryTop
PM and DM are the most common idiopathic inflammatory myopathies in adults (although sporadic inclusion body myositis [sIBM] is more common in older men and women aged >50 years). sIBM will be only briefly considered in this chapter, as it is still unclear whether the associated T-cell–mediated inflammation is a primary factor or secondary phenomenon due to an underlying degenerative etiology. PM and DM affect women twice as frequently as men, with an incidence of between 1 to 8 cases per 1,000,000 per year (inclusion body myositis is ~3 times more common in men than women). PM and DM may occur at any age; however, the onset of DM is most often between the ages of 10 to 15 years for the juvenile form and 35 to 65 years for the later-onset form. PM is much more common in adulthood and is rarely diagnosed in those <20 years of age. PM can be idiopathic but is often associated with features of another connective tissue disease and is sometimes thus referred to as overlap myositis. The onset is usually acute (lasting a few days) to subacute (weeks) but can rarely have an indolent course over months to years.
1. General symptoms are fatigue, fever, and weight loss.
2. Symptoms of muscle involvement:
1) Usually symmetric weakness of muscles of the shoulder girdle, hip flexors, neck flexors, and back extensors can lead to problems with such tasks as standing up from a sitting position, lifting heavy objects, brushing hair, lifting a head off a pillow or climbing stairs. The muscles may be tender and painful. Muscles may eventually undergo atrophy (especially myositis associated with antibodies against signal recognition particle [SRP]) and contribute to weight loss.
2) Respiratory muscle weakness may result in shortness of breath on exertion and occasionally respiratory failure.
3) Weakness of the muscles of the throat and upper esophagus are less common but can cause dysphagia and very rarely nasal dysarthria.
4) Involvement of extraocular muscles resulting in ophthalmoparesis and/or ptosis is so rare that its presence should warrant an investigation into alternate diagnoses.
3. Cutaneous manifestations occur in DM and PM. Their onset and severity may not be correlated with muscle involvement. Cutaneous manifestations may precede myositis or be the only sign of the disease in some cases of DM (clinically amyopathic dermatomyositis [CADM]; dermatomyositis sine myositis). CADM is often associated with antibodies against melanoma differentiation-associated gene 5 (MDA5/CADM-140). Erythematous lesions may be frequently accompanied by pruritus and/or skin photosensitivity.
1) DM: Periorbital violaceous erythema (heliotrope rash; Figure 18.19-1) is a pathognomonic DM sign occurring in 30% to 60% of patients and is occasionally accompanied by lid edema (more common in juvenile DM). Erythematous rash on the upper chest (the V sign), on the neck and shoulders (the shawl sign; Figure 18.19-2), or on the lateral thighs and hips (the holster sign) may also develop.
2) DM: Gottron rash/sign is seen in ~70% of patients and is more commonly seen in DM (see Figure 1.15-5). It consists of erythematous/violaceous eruptions on the extensor surfaces of finger joints (most frequently the proximal and distal interphalangeal and metacarpophalangeal joints with sparing between the joints but occasionally seen on wrists, elbows, knees, and ankles). Gottron papules are rarer and are a raised erythematous/violaceous coalescence of the Gottron rash, but can occur on the lateral side of the fingers.
3) Other features seen in PM and DM: “Mechanic’s hands” (Figure 18.19-3), which is a thickening, desquamation, and cracking of the skin on the palmar surfaces of the fingers and the hand, more common in antisynthetase syndrome and in those with interstitial lung disease (see below); erythema and periungual edema of the nail folds with petechiae and dilated capillary loops/telangiectasias; trophic ulcers due to coexisting cutaneous vasculitis; generalized erythroderma; inflammation of the subcutaneous tissue (panniculitis); livedo reticularis; and focal alopecia without scarring.
4. Cardiac manifestations are found with extensive testing in up to 70% of patients, usually in the form of tachycardia or bradycardia, but are rarely symptomatic. Signs and symptoms of heart failure are similarly rare.
5. Pulmonary manifestations: Symptoms of interstitial lung disease are seen in up to 30% of patients, predominantly in those with antisynthetase syndrome. Symptoms are mainly dry cough and worsening dyspnea progressing to chronic respiratory failure. In rare cases patients with dysphagia can develop aspiration pneumonia.
6. Gastrointestinal (GI) manifestations: Dysphagia is the most common feature, but patients may also have features of impaired esophageal, gastric, and intestinal motility, including gastroesophageal reflux. Dysphagia may persist after the proximal muscle weakness and CK elevation have resolved and may lead to the need for a feeding tube. In severe cases ulcerations and bleeding may occur. Consequently, proton pump inhibitors are often given simultaneously with glucocorticoids if the patient is symptomatic or has endoscopically determined esophagitis.
7. Joint manifestations: Symptoms of arthritis without erosions, or arthralgia, with a predilection for peripheral joints, mainly of the hands (in 20%-50% of patients). These features are more common in those with overlap myositis (ie, myositis associated with another connective tissue disorder, mixed connective tissue disease [MCTD], systemic lupus erythematosus [SLE], scleroderma).
8. Calcifications in the subcutaneous tissue, skeletal muscles, fasciae, and tendons are very rare with early treatment but may be of considerable size if treatment is delayed, especially in juvenile DM and some cases of PM with positive antibodies against SRP and antisynthetase antibody.
9. Raynaud phenomenon is seen in 10% to 15% of patients.
DiagnosisTop
1. Laboratory tests:
1) Blood tests: Elevated plasma levels of muscle enzymes (creatine kinase [CK], aspartate aminotransferase [AST], alanine aminotransferase [ALT], lactate dehydrogenase [LDH], aldolase, myoglobin), hypergammaglobulinemia, increased erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels.
2) Immunologic tests: Autoantibodies are seen in 40% to 80% of patients and include antinuclear antibodies (ANAs); antibodies to double-stranded DNA (dsDNA), Ro, La, U1/U2 ribonucleoprotein (RNP), Ku, Sm; antibodies specifically associated with PM/DM (ie, antibodies to aminoacyl-tRNA synthetase [most commonly Jo-1]); and antibodies to SRP, transcription intermediary factor 1gamma/alpha (TIF1gamma/alpha, p155/140), MJ/nuclear matrix protein 2 (NXP-2), small ubiquitin-like modifier-1 (SUMO-1) activating enzyme (SAE), Mi-1, Mi-2, MDA5, PL7, PM/Scl75, PM/Scl100, Ku, Ej, and Oj. Other than the ANAs, most of the tests for the antibodies listed above are only available in specialty clinics but can be helpful from both a diagnostic and sometimes a prognostic perspective (see below).
2. Electromyography (EMG) usually shows spontaneous activity (fibrillations, positive sharp waves, pseudomyotonic discharges; more common in PM) with small, brief, early recruiting potentials in both PM and DM.
3. Histology:
1) Muscle biopsy typically shows a dense T-cell–mediated inflammatory reaction with inflammatory cells invading nonnecrotic fibers in PM. DM changes include perifascicular atrophy and B-cell infiltration often in the epimysium, perimysium, and/or blood vessels, with immunohistochemistry showing membrane attack complex deposits around blood vessels. Electron microscopy frequently shows inflammatory cells in both DM and PM, but tubuloreticular inclusions in blood vessels are more commonly seen in DM and are thought to reflect a viral etiology and presence of interferon gamma.
2) Lung (surgical) biopsy may reveal interstitial lung disease (various types have been observed), but this is rarely required, because typical features on chest radiography or high-resolution computed tomography (HRCT) and clinical symptoms are characteristic (the ground-glass appearance on chest radiographs and impaired carbon monoxide diffusion capacity [DLCO]).
4. Imaging studies: In the active phase, muscle magnetic resonance imaging (MRI) shows an increased signal intensity on fat-saturated, gadolinium-enhanced, T1-weighted, T2-weighted, or short tau inversion recovery (STIR) images; however, the findings are not pathognomonic for PM or DM. In the chronic phase, T1-weighted images may show atrophy of adipose tissue, muscle, or both. Imaging studies may facilitate the choice of an optimal site for muscle biopsy. Chest radiographs and HRCT may reveal interstitial lesions. Radiographs of bones and joints may reveal numerous calcifications, mainly within the subcutaneous tissue and muscles, as well as features of osteoporosis.
5. Electrocardiography (ECG) usually shows nonspecific ST-segment and T-wave abnormalities, sinus tachycardia, and less frequently first- or second-degree atrioventricular block.
Diagnostic criteria: Table 18.19-1.
Differential diagnosis should include:
1) Idiopathic inflammatory myopathies other than PM/DM: Myositis can be a presenting manifestation of several systemic connective tissue diseases, including SLE, rheumatoid arthritis, MCTD, Sjögren syndrome, and scleroderma (overlap myositis syndrome). The characteristic autoantibodies are important adjuncts to the history and physical examination in the diagnosis of these entities.
2) Necrotizing autoimmune myopathies often clinically resemble PM and are most commonly seen in association with statin use, with up to 50% showing T-cell infiltrates in addition to necrosis at some point in the disease course. These myopathies show canonical features of PM (usually there is no rash, as seen in DM) with high CK levels, classical EMG findings of PM, proximal weakness, and inflammation on biopsy. Most (>90%) patients with acute statin-induced rhabdomyolysis show a good clinical response to discontinuing the offending drug; however, if the CK elevation and weakness persist, it is likely that the patient has developed autoantibodies against HMG-CoA-reductase (HMGCR).Evidence 1Moderate Quality of Evidence (moderate confidence that we know true effects of the intervention). Quality of Evidence lowered due to indirectness (likely severely affected patients). Nazir S, Lohani S, Tachamo N, Poudel D, Donato A. Statin-Associated Autoimmune Myopathy: A Systematic Review of 100 Cases. J Clin Rheumatol. 2017 Apr;23(3):149-154. doi: 10.1097/RHU.0000000000000497. Review. PubMed PMID: 28277343. This group of statin myopathies invariably requires treatment with prednisone and, if needed, a disease-modifying agent to allow for glucocorticoid sparing (methotrexate being the most common). Necrotizing myopathies can also be associated with systemic connective tissue diseases, viral infections (eg, HIV), or malignancy. History, examination, and specific blood tests usually differentiate these entities from idiopathic PM.
3) sIBM is often mistaken for PM, and vice versa, in that sIBM shows hyperCKemia, T-cell infiltrates on biopsy, and EMG findings overlapping with PM. The differentiating features for sIBM are specific weakness of deep finger flexors as well as disproportionate weakness/atrophy of the quadriceps muscles and dysphagia in ~70% of cases, and often a very slow onset of symptoms that upon retrospect started many years earlier. Furthermore, the muscle biopsy in sIBM often shows the presence of inclusion bodies and/or mitochondrial alterations by modified Gomori trichrome staining or electron microscopy. The inclusion bodies can be seen in hereditary forms of inclusion body myositis (hIBM) but the clinical features are distinct (eg, the most common hIBM is due to mutations in the GNE gene and has an insidious onset of distal lower extremity weakness, with quadriceps sparing often starting in teenage years). Although CK levels in inclusion body myositis are often lower than those seen in PM or DM, a modest CK elevation (<1000 IU) cannot be used to exclude PM or DM. In contrast to PM/DM, there is a 3:1 male to female ratio in sIBM, and the disease is more common in those aged >50 years, with no juvenile cases. Unlike in PM and DM, there is no clinically meaningful response to immune suppression (IgG, prednisone).
4) Other myopathies: Table 18.19-2.
Search for malignancy: see Follow-Up, below.
TreatmentTop
1. Glucocorticoids: Oral prednisone 1 mg/kg/d (maximum, 60 mg). In patients with an acute onset or a severe course of the disease, IV methylprednisolone 0.5 to 1 g may be used as the initial treatment for 3 days in severe cases and in proportionately lower doses in less severe cases. After observing an improvement in the patient’s condition (ie, increase in muscle strength and a substantial reduction in muscle enzymes; CK is all that is needed to follow the muscle damage; this is usually seen within 6 weeks), taper the dose, for instance, by ~10 mg a month down to 10 mg/d, and then taper more slowly (2.5 mg per month) until 5 mg/d, at which point tapering can continue at 1 mg/mo. If the CK level shows a consistent pattern of rising at a given dose and the patient starts to experience the return of symptoms, the dose should be increased by ≥50% and disease-modifying therapy should be introduced. If the prednisone dose is ≤5 mg/d, monotherapy can be considered due to the low risk of long-term adverse effects.
2. In case of either no satisfactory improvement within 6 weeks of starting treatment, a return of symptoms, or an increasing CK level (as described above), one should add a disease-modifying agent:
1) Methotrexate: Administer 10 to 25 mg/wk orally or parenterally (IM or subcutaneously) once a week, with folate 10 mg/wk or folinic acid (the same dose) the day after, for GI protection. Therapeutic effects may be observed up to 3 months after starting treatment. Start with a dose adjusted to the severity of the disease, for instance, 10 to 15 mg once weekly, and if the symptoms do not resolve, titrate the dose up by 5 mg every month. Monitor the complete blood count (CBC), gamma-glutamyl transferase (GGT), bilirubin, and ALT monthly for 6 months and every 2 months thereafter. After a cumulative dose of 5 g, a referral to a gastroenterologist for consideration of transient elastography (FibroScan) or liver biopsy may be made.
2) Azathioprine: Therapeutic effects may be observed up to 4 months after starting treatment, and an increase in the mean corpuscular volume (MCV) >100 fL is often reflective of the optimal dose (1.5-2.5 mg/kg/d). Monitor the CBC, GGT, bilirubin, and ALT monthly for 6 months and every 2 months thereafter. About 7% of patients will develop a flu-like illness within days of starting the agent (avoid the use of azathioprine with allopurinol or other xanthine oxidase inhibitors).
3) Intravenous immunoglobulin (IVIG): This is often effective in combination with methylprednisolone (250-1000 mg/d) in severe disease and has been shown to be effective in lowering the steroid dose in both DM and PM.Evidence 2Moderate Quality of Evidence (moderate confidence that we know true effects of the intervention). Quality of Evidence lowered due to the observational nature of the study and increased due to the size of the effect. Cherin P, Pelletier S, Teixeira A, et al. Results and long-term follow-up of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum. 2002 Feb;46(2):467-74. PubMed PMID: 11840450. Miyasaka N, Hara M, Koike T, Saito E, Yamada M, Tanaka Y; GB-0998 Study Group. Effects of intravenous immunoglobulin therapy in Japanese patients with polymyositis and dermatomyositis resistant to corticosteroids: a randomized double-blind placebo-controlled trial. Mod Rheumatol. 2012 Jun;22(3):382-93. doi: 10.1007/s10165-011-0534-4. Epub 2011 Oct 5. PubMed PMID: 21971943; PubMed Central PMCID: PMC3375426. Elovaara I, Apostolski S, van Doorn P, et al; EFNS. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurologic diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurologic diseases. Eur J Neurol. 2008 Sep;15(9):893-908. doi: 10.1111/j.1468-1331.2008.02246.x. Erratum in: Eur J Neurol. 2009 Apr;16(4):547. PubMed PMID: 18796075. Lünemann JD, Nimmerjahn F, Dalakas MC. Intravenous immunoglobulin in neurology--mode of action and clinical efficacy. Nat Rev Neurol. 2015 Feb;11(2):80-9. doi: 10.1038/nrneurol.2014.253. Epub 2015 Jan 6. Review. PubMed PMID: 25561275. Use a dose of 1 g/kg/d twice in 2 to 4 days or 0.4 g/kg/d for 5 consecutive days and then once a month for 3 to 7 months.
4) Cyclosporine (INN ciclosporin) in doses 2.5 to 5 mg/kg/d with prednisone was shown to be superior to prednisone alone. Cyclosporine is associated with a greater number of infections compared to prednisone alone. Renal function and blood pressure must be carefully monitored with cyclosporine use.
5) Cyclophosphamide 1 g IV once a month or 1 to 3 mg/kg orally once daily. This agent is generally only used in patients with severe interstitial pulmonary lesions not responding to standard therapy or in those with severe vasculitis.
6) Mycophenolate mofetil 2 to 3 g/d in case methotrexate, azathioprine, and cyclosporine are poorly tolerated or ineffective.
7) Hydroxychloroquine 200 mg bid or chloroquine 250 mg bid are indicated in patients with refractory cutaneous lesions.
8) Biologic agents: A variety of biologics have been tried primarily in juvenile DM, but their long-term safety and efficacy has not been determined. In the authors’ experience, these agents are very rarely indicated (rituximab, abatacept, anti–tumor necrosis factor alpha drugs, and tocilizumab).
9) Combination therapy: Most patients are treated with a combination of a glucocorticoid and IVIG or another disease-modifying agent. In severe treatment-resistant cases one may consider the combination of methotrexate and azathioprine or one of available combinations with mycophenolate.
Exercise and physiotherapy are important measures to prevent issues and to restore physical function. In patients with acute disease mainly passive movements and stretching are recommended to prevent contractures. When subjective strength is improving and the CK level is closer to normal, the patient can start endurance and resistance exercises, initially at a low level and gradually increasing the intensity and duration as tolerated. In general, the suggested aerobic intensity is ≤65% of the maximal aerobic power; for resistance exercise, the number of suggested repetitions is >12; and the suggested intensity is ≤65% of the one-repetition maximum.
Follow-UpTop
Follow-up should involve periodic evaluation of muscle strength and plasma CK levels; for patients receiving disease-modifying agents, the CBC, liver function/damage tests (bilirubin, GGT, ALT; note that ALT will follow CK levels and is only relevant if disproportionate to CK); and in patients with interstitial lung disease, HRCT, pulmonary function tests, and the 6-minute walk test.
Assessment and monitoring for malignancy is important but need not be exhaustive (chest radiographs in smokers or those with respiratory symptoms, occult blood/colonoscopy [especially in those aged >50 years], mammography/breast examination, Papanicolaou test in women not immunized against human papillomavirus, abdominal and pelvic ultrasound in those with abdominal symptoms and women with altered periods, prostate examination [clinical and with prostate-specific antigen test] in men aged >50 years). The risk of cancer is highest within the first 3 years of the diagnosis of PM/DM but continues to be high even after 5 years. Other factors associated with an increased risk of malignancy include age >50 years, DM, severe skin involvement, and the presence of anti-155/140 kD (TIF1gamma) antibodies.
PrognosisTop
The 5-year mortality rate for treated patients is between 20% and 35%, but for cancer-associated myositis the overall mortality rate (related to primary disease, drugs, or myositis complications) can be as high as 75%. A less favorable prognosis is also associated with advanced age and involvement of internal organs, particularly lungs, and the presence of antibodies to SRP. A more favorable prognosis is seen in PM cases associated with the Mi-2 antibody. The risk of malignancy is 4-fold to 6-fold greater than for aged-matched controls in adult patients with DM and 1.5-fold to 2-fold greater in patients with PM (including the increased risk of ovarian, breast, lung, intestinal, nose and throat, pancreatic, and bladder cancers as well as non-Hodgkin lymphomas).
Tables and FiguresTop
|
1. Symmetric progressive muscle weakness affecting the muscles of the shoulder and hip 2. Histologic findings typical of myositis (see text) 3. Increased plasma creatine kinase levels 4. Electromyographic features of an irritative myopathy (active changes and myopathic potentials) 5. Typical cutaneous manifestations: Gottron sign, heliotrope rash, or rash on the anterior chest and shoulders | |||
|
Diagnosis |
Definite |
Probable |
Possible |
|
Number of symptoms | |||
|
PM |
4 |
3 |
2 |
|
DM |
3 or 4 |
2 |
1 |
|
Plus typical cutaneous manifestations | |||
|
Adapted from N Engl J Med. 1975;292(7):344-7. | |||
|
DM, dermatomyositis; PM, polymyositis. | |||
|
Endocrine disorders |
Hypothyroidism, hypercortisolemia, thyrotoxicosis, hypoparathyroidism, hyperparathyroidism Testing: Endocrine blood testing |
|
Metabolic disorders |
GSDs (primarily McArdle disease [type V] but also III, VII, and XV), lysosomal storage disease (Pompe, previously known as GSD II), mitochondrial myopathy (TK2, MELAS, and other mtDNA mutations), primary systemic carnitine deficiency Testing: Specific genetic testing (including mtDNA sequencing or nuclear-encoded gene testing, dry blood spot for Pompe disease, muscle histology and enzymatic testing (ie, PFK [GSD VII] and phosphorylase [GSD V]) |
|
Muscular dystrophy |
Most common dystrophies to be mistaken for PM are Miyoshi myopathy due to dysferlin (DYSF) or ANO5 mutations, dystrophinopathy (mainly Becker), other limb girdle MDs (eg, sarcoglycanopathy, caveolin-3). Because nearly every one of >70 types of MD will meet probable criteria for PM (proximal weakness, high CK, myopathic EMG), careful history and examination are essential (medial calf atrophy is characteristic of Miyoshi myopathy and not PM/DM), muscle biopsy (inflammation is common as a secondary finding in MD but invasion is of necrotic fibers unlike in PM where they are nonnecrotic; immunohistochemistry can be classic: patchy or absent dystrophin staining in Becker) Testing: Immunohistochemistry on muscle biopsy samples and specific genetic testing |
|
Nerve disorders |
Spinal muscular atrophy type III (EMG is usually characteristic, CK is normal or minimally elevated), amyotrophic lateral sclerosis (weakness is usually asymmetric but CK can be elevated [usually <1000 IU], EMG shows active changes but recruitment is different than in PM [reduced and large, long vs small duration, brief and early recruiting in PM/DM]) Testing: Genetic testing for SMA; history, examination, and EMG for ALS (El-Escorial criteria) |
|
Neuromuscular junction |
LEMS can present with proximal weakness but EMG is not characteristic of PM/DM and CK is usually not elevated. Diagnosis is established with incremental CMAP response post contraction and presence of antibodies to P/Q-type voltage-gated calcium channels. MG usually presents with ocular and bulbar symptoms and only rarely proximal weakness; CK is not usually elevated and EMG is normal. Diagnosis is established by single-fiber EMG, decremental response of CMAP to repetitive stimulation, and presence of specific antibodies (antiacetylcholine receptor, anti-MuSK) Testing: EMG is suggestive but presence of antibodies is very specific |
|
Infections |
Viral, bacterial, parasitic (toxoplasmosis, trichinosis) Testing: Virology, bacterial culture, and PCR-based testing |
|
Electrolyte disturbances |
Decreased or increased plasma levels of sodium, potassium, calcium, magnesium, or phosphate Testing: Electrolyte analysis in blood |
|
Granulomatous diseases |
Sarcoidosis Testing: Noncaseating granulomas in muscle biopsy, low ACE levels in blood |
|
Drugs |
Lipid-lowering agents (statins, rarely fibrates), antimalarial agents (chloroquine and hydroxychloroquine), amiodarone, quinidine, cimetidine, cyclosporine (INN ciclosporin), danazol, D-penicillamine, interferon alpha, interleukin 2, colchicine, epsilon-aminocaproic acid, phenylbutazone, phenytoin, glucocorticoids (common: type 2 muscle fiber atrophy, especially with doses >10 mg), hydralazine, levodopa, penicillin, procainamide, rifampicin, sulfonamides, L-tryptophan, valproic acid, vincristine, some anti-HIV drugs (zidovudine, ddI, ddC) |
|
Toxic substances |
Acute exposure to carbon monoxide, alcohol, heroin, cocaine; history and specific testing will easily differentiate these from PM/DM |
|
ACE, angiotensin-converting enzyme; ALS, amyotrophic lateral sclerosis; CK, creatine kinase; CMAP, compound muscle action potential; DM, dermatomyositis; EMG, electromyography; GSD, glycogen storage disease; INN, international nonproprietary name; LEMS, Lambert-Eaton myasthenic syndrome; MD, muscular dystrophy; MELAS, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; MG, myasthenia gravis; mtDNA, mitochondrial DNA; MuSK, muscle-specific kinase; PCR, polymerase chain reaction; PFK, phosphofructokinase; PM, polymyositis; SMA, spinal muscular atrophy; TK2, thymidine kinase 2. | |
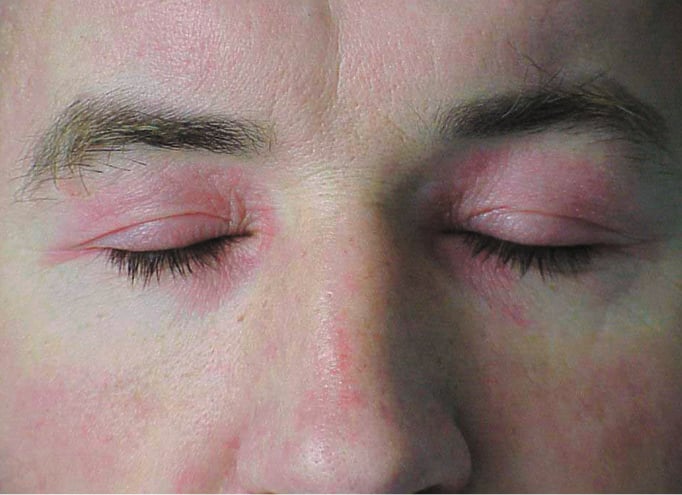
Figure 18.19-1. Polymyositis/dermatomyositis: periorbital heliotrope rash.
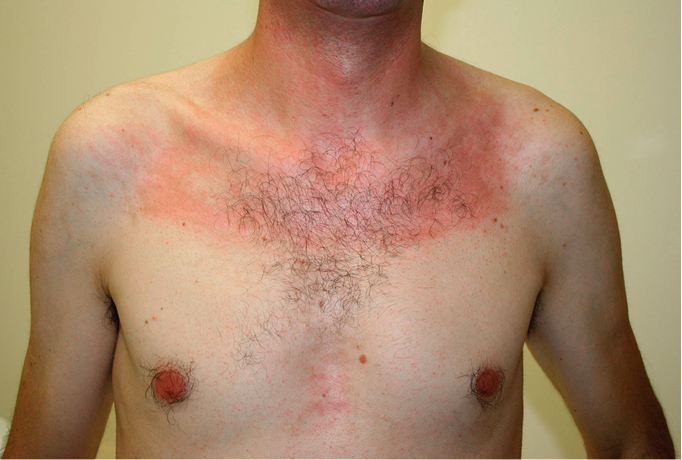
Figure 18.19-2. Polymyositis/dermatomyositis: Shawl sign and V-sign. Photograph courtesy of Dr Mark Tarnopolsky.
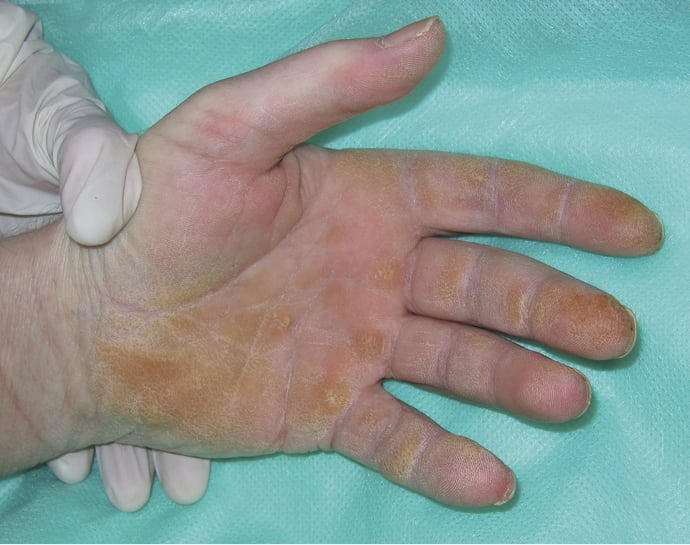
Figure 18.19-3. “Mechanic’s hands.”