 English
English
 Español
Español
 українська
українська

Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022 May 3;79(17):1757-1780. doi: 10.1016/j.jacc.2021.12.011. Epub 2022 Apr 1. PMID: 35379504.
McDonald M, Virani S, Chan M, et al. CCS/CHFS Heart Failure Guidelines Update: Defining a New Pharmacologic Standard of Care for Heart Failure With Reduced Ejection Fraction. Can J Cardiol [Internet]. 2021 Apr;37(4):531-546. doi: 10.1016/j.cjca.2021.01.017. PMID: 33827756.
Bozkurt B, Coats AJ, Tsutsui H, et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J Card Fail. 2021 Mar 1;S1071-9164(21)00050-6. doi: 10.1016/j.cardfail.2021.01.022. PMID: 33663906.
McDonagh TA, Metra M, Adamo M, et al; ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021 Sep 21;42(36):3599-3726. doi: 10.1093/eurheartj/ehab368. Erratum in: Eur Heart J. 2021 Dec 21;42(48):4901. PMID: 34447992.
Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020 Dec 22;76(25):e159-e240. doi: 10.1016/j.jacc.2020.08.045. Epub 2020 Nov 20. PMID: 33229116.
Cadrin-Tourigny J, Bosman LP, Wang W, et al. Sudden Cardiac Death Prediction in Arrhythmogenic Right Ventricular Cardiomyopathy: A Multinational Collaboration. Circ Arrhythm Electrophysiol. 2021 Jan;14(1):e008509. doi: 10.1161/CIRCEP.120.008509. Epub 2020 Dec 9. PMID: 33296238; PMCID: PMC7834666.
Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019 Nov;16:e301-e372. doi: 10.1016/j.hrthm.2019.05.007. Epub 2019 May 9. PMID: 31078652.
Cadrin-Tourigny J, Bosman LP, Nozza A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2019 Jun 14;40(23):1850-1858. doi: 10.1093/eurheartj/ehz103. PMID: 30915475; PMCID: PMC6568197.
Corrado D, Wichter T, Link MS, et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Circulation. 2015 Aug 4;132(5):441-453. doi: 10.1161/CIRCULATIONAHA.115.017944. Epub 2015 Jul 27.
Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010 Apr 6;121(13):1533-41. doi: 10.1161/CIRCULATIONAHA.108.840827. Epub 2010 Feb 19. PMID: 20172911; PMCID: PMC2860804.
Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008 Jan;29(2):270-6. Epub 2007 Oct 4. PMID: 17916581.
Definition, Etiology, PathogenesisTop
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetic disease involving mainly the right ventricle. It is caused by the gradual replacement of myocardial fibers by fatty and fibrous tissue, particularly in the right ventricular inflow, outflow, and apex, which leads to a propensity to ventricular arrhythmias and sudden cardiac death (SCD). Morphologic and functional changes can also occur in the left ventricle (LV), producing a phenotype similar to dilated cardiomyopathy. Due to the potential to involve the left ventricle, experts have suggested that the condition be more appropriately named arrhythmogenic cardiomyopathy (ACM), and ARVC be reserved for the subtype that is restricted to the right ventricle. The term ACM is now generally viewed to collectively refer to a heterogeneous group of presumed genetic cardiomyopathies manifesting with an arrhythmic risk that is out of proportion with the degree of ventricular dysfunction present. In this context, a left ventricular ejection fraction (LVEF) threshold of <35% for the consideration of primary prevention implantable cardioverter-defibrillator (ICD) therapy may not be sufficient to provide optimal protection against SCD.
Causes: Pathogenic rare genetic variants, which are usually autosomal dominant, although penetrance is variable. The majority of genetic culprits implicated in ARVC are constituents of the cardiac desmosome.
Clinical Features and Natural HistoryTop
1. History: ARVC most commonly presents between the third and fourth decades of life, with a possible predilection for males. The first symptom may be palpitations, syncope, or SCD secondary to ventricular arrhythmia. Atrial arrhythmias, including atrial fibrillation, may also develop. Heart failure, most commonly right sided, may also occur in ARVC; however, it usually develops at later stages of the disease.
2. Risk factors for SCD include young age at clinical presentation, history of syncope, cardiac arrest or hemodynamically significant ventricular tachycardia, LV involvement, and significant right ventricular damage. Risk stratification for ARVC and decisions regarding the appropriate timing for insertion of a primary prevention ICD may be challenging given that the risk of ventricular arrhythmias depends on the collective clinical and genetic profile of the patient rather than on a single clinical feature. In this context, a multivariable model that incorporates multiple clinical features has been developed to predict arrhythmia risk and guide ICD decision-making (arvcrisk.com).
3. Symptoms include palpitations, presyncope, and syncope. In advanced disease symptoms of right-sided, left-sided, or biventricular heart failure may develop.
DiagnosisTop
1. Electrocardiography (ECG): The most common finding is inverted T waves in the anterior precordial leads, often in V1 through V3, although T-wave inversion may extend through V6. An epsilon wave, defined as a low-frequency late potential that is offset from the QRS, is a classic finding in ARVC, although it is present only in a minority of patients. It is reflective of a region of slow conduction within the right ventricle and is usually present in V1, V2, or both. Although often suggestive of ARVC, it is not pathognomonic, as it may develop in the presence of any condition that leads to remarkable right ventricular scarring and associated slow conduction, with cardiac sarcoidosis being another prominent example. When present, ventricular arrhythmias most often have a left bundle branch block (LBBB) morphology of the QRS complex, consistent with their arising from the right ventricle.
2. Echocardiography (Figure 3.6-1) may reveal impaired right ventricular systolic function and enlargement of the right ventricle. Focal wall motion abnormalities of the right ventricle, including aneurysms, may also be observed.
3. Magnetic resonance imaging (MRI) may also reveal right ventricular dilation and reduced systolic function. Use of late gadolinium enhancement may show fibrosis in the right ventricular wall. Fatty replacement of the right ventricular myocardium, although a classic finding, is observed relatively infrequently.
4. Genetic testing may identify the causative mutation and add to family screening.
ARVC is diagnosed on the basis of the Revised Task Force Criteria. The diagnostic Task Force Criteria for ARVC consist of a constellation of structural alterations (most often identified on imaging studies), electrocardiographic findings (both depolarization and repolarization abnormalities), cardiac arrhythmias, and family history (including identification of a pathogenic variant known to cause the condition). Features from these broad categories are classified as major and minor criteria. Depending on the number of major and minor criteria, an individual can be assigned a definite, borderline, or possible diagnosis of ARVC.
ARVC should be differentiated from idiopathic right ventricular outflow tract ventricular tachycardia, cardiac sarcoidosis, myocarditis, Brugada syndrome, right ventricular myocardial infarction, and dilated cardiomyopathy.
TreatmentTop
1. Symptomatic treatment targets arrhythmia and includes mainly sotalol, beta-blockers, or amiodarone.
2. Radiofrequency ablation is used in case of intolerance or ineffectiveness of antiarrhythmic agents in patients with life-threatening arrhythmia. Owing to ARVC pathophysiology arising within the epicardium and progressing inwards through the myocardium to the endocardium, endocardial ablation in isolation is often insufficient to effectively suppress ventricular arrhythmias and epicardial ablation is additionally required.
3. ICDs are used to prevent SCD in patients with severe ventricular arrhythmia and syncope. Decision-making regarding timing of primary prevention ICD insertion is nuanced and can be guided by the web-based risk calculator at arvcrisk.com.
4. Patients with heart failure (right sided, left sided, or biventricular) require guideline-directed medical therapy.
5. Patients with end-stage heart failure, refractory ventricular arrhythmias, or both should be considered for heart transplantation.
FiguresTop
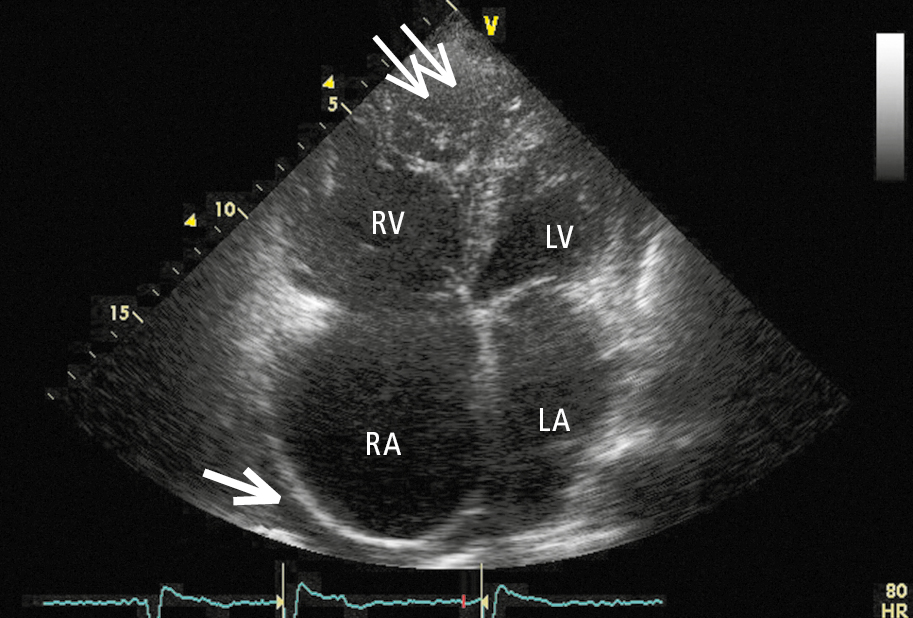
Figure 3.6-1. Transthoracic echocardiography (TTE) of a patient with arrhythmogenic right ventricular cardiomyopathy (ARVC). A modified apical 4-chamber view showing diffuse right ventricular injury, ample trabeculation at the apex (thin arrows), significant right atrial dilatation, and separation of pericardial layers due to pericardial fluid accumulating behind the right atrium (thick arrow). LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. Figure courtesy of Dr Marek Konka.