 English
English
 Español
Español
 українська
українська

Khan AA, Bilezikian JP, Brandi ML, et al. Evaluation and Management of Hypoparathyroidism Summary Statement and Guidelines from the Second International Workshop. J Bone Miner Res. 2022 Dec;37(12):2568-2585. doi: 10.1002/jbmr.4691. Epub 2022 Nov 14. PMID: 36054621.
Yao L, Hui X, Li M, et al. Complications, Symptoms, Presurgical Predictors in Patients With Chronic Hypoparathyroidism: A Systematic Review. J Bone Miner Res. 2022 Dec;37(12):2642-2653. doi: 10.1002/jbmr.4673. Epub 2022 Nov 14. PMID: 36375810.
Khan AA, Guyatt G, Ali DS, et al. Management of Hypoparathyroidism. J Bone Miner Res. 2022 Dec;37(12):2663-2677. doi: 10.1002/jbmr.4716. Epub 2022 Oct 31. PMID: 36161671.
Alalawi Y, El Werfalli R, Abu Alrob H, et al. MON-LB091 An Overview of the Etiology, Clinical Manifestations, Management Strategies, and Complications of Hypoparathyroidism from the Canadian National Hypoparathyroidism Registry. J Endocr Soc. 2019 Apr 30;3(Suppl 1):MON-LB091. doi: 10.1210/js.2019-MON-LB091. PMCID: PMC6551102.
Khan AA, Koch C, Van Uum SHM, et al. Standards of Care for Hypoparathyroidism in Adults. Eur J Endocrinol. 2018 Dec 1. pii: EJE-18-0609.R1. doi: 10.1530/EJE-18-0609. [Epub ahead of print] PubMed PMID: 30540559; PubMed Central PMCID: PMC6365672.
Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N Engl J Med. 2018 Mar 22;378(12):1132-1141. doi: 10.1056/NEJMra1713301. Review. PMID: 29562162; PMCID: PMC6007870.
Brandi ML, Bilezikian JP, Shoback D, et al. Management of Hypoparathyroidism: Summary Statement and Guidelines. J Clin Endocrinol Metab. 2016 Jun;101(6):2273-83. doi: 10.1210/jc.2015-3907. Epub 2016 Mar 4. Review. PMID: 26943719.
Mitchell DM, Regan S, Cooley MR, et al. Long-term follow-up of patients with hypoparathyroidism. J Clin Endocrinol Metab. 2012 Dec;97(12):4507-14. doi: 10.1210/jc.2012-1808. Epub 2012 Oct 5. PMID: 23043192; PMCID: PMC3513540.
Definition, Etiology, PathogenesisTop
Hypoparathyroidism is a condition associated with hypocalcemia and hyperphosphatemia in the presence of low or inappropriately normal parathyroid hormone (PTH) levels (see Figure 6.6-2). It is associated with significant symptoms of hypocalcemia as well as long-term complications of inadequate PTH levels, hypocalcemia, and hyperphosphatemia.
Causes (Table 6.6-1):
1) Neck surgery is the most common cause of primary hypoparathyroidism, accounting for 75% to 80% of all cases. During neck surgery the parathyroid glands may be damaged or deprived of their blood supply. Patients with transient hypoparathyroidism after surgery may recover; however, if the serum calcium level remains low for ≥12 months, the diagnosis of chronic hypoparathyroidism is confirmed.
2) Nonsurgical causes of hypoparathyroidism account for 20% to 25% of cases:
a) Autoimmune hypoparathyroidism is the most common cause of nonsurgical primary hypoparathyroidism. It may occur in isolation or as part of autoimmune polyendocrine syndrome type 1 (APS-1). APS-1 is caused by sequence variations in the AIRE (autoimmune regulator) gene. The cardinal features of APS-1 include adrenal insufficiency, hypoparathyroidism, and mucocutaneous candidiasis. At least 80% of patients with APS-1 have hypoparathyroidism. Individuals with APS-1 should be followed up to ensure that other major or minor features of APS do not develop. In isolated autoimmune hypoparathyroidism, activating antibodies directed to the calcium-sensing receptor (CaSR) cause decreased PTH secretion. Currently, there is no gold standard test for CaSR antibodies, which makes it challenging to diagnose and address the prevalence of the disease.
b) Genetic causes can be divided into 2 main categories: syndromic and nonsyndromic. DiGeorge syndrome occurs due to a deletion at the site of chromosome 22q11.2, which leads to a developmental defect of the pharyngeal pouches and results in hypoparathyroidism in addition to several immunologic, cardiac, renal, and developmental anomalies. Autosomal dominant hypocalcemia (ADH) type 1 is a nonsyndromic cause that occurs due to an activating (gain of function) sequence variation of the CASR gene. This results in hypocalcemia due to decreased synthesis and secretion of PTH.
c) Infiltrative causes include destruction of the parathyroid glands secondary to granulomatous infiltration (eg, Riedel thyroiditis, amyloidosis, sarcoidosis). Metastatic cancer cells infiltrate the parathyroid gland leading to hypoparathyroidism. Wilson disease causes hypoparathyroidism due to copper infiltration and destruction of parathyroid glands. Hemochromatosis is an inherited disorder resulting in increased iron deposition in various organs, including the parathyroid glands, which leads to parathyroid gland dysfunction.
d) Radiation destruction can also be a cause, although high doses of ionizing radiation exposure have been rarely associated with hypoparathyroidism.
e) Transient hypoparathyroidism can be seen in the presence of severe burn injuries and acute illness.
f) Functional hypoparathyroidism is caused by hypomagnesemia as well as hypermagnesemia, both of which impair parathyroid function (Table 6.6-2).
g) Maternal hyperparathyroidism can result in suppressed parathyroid function in infants exposed to hypercalcemia in utero.
h) Idiopathic hypoparathyroidism is confirmed if the cause of hypoparathyroidism is not identified after laboratory, genetic, or clinical evaluation.
Two additional clinical states have traditionally carried “hypoparathyroidism” as part of their name but are either distinct or not a disease entity. Pseudohypoparathyroidism (PHP) is defined as target organ resistance to PTH. PHP is characterized by elevated PTH levels in association with low serum calcium and high phosphate levels. Secondary hypoparathyroidism is not a disease state but a physiologic response, where PTH levels are low in response to a primary process that has caused hypercalcemia, not hypocalcemia (see Hypercalcemia).
Clinical Features and Natural HistoryTop
1. Symptoms of hypocalcemia include muscle cramping; twitching; numbness and tingling in the face, hands, or feet; depression or irritability; seizures; bradyarrhythmia; wheezing; and laryngospasm (Table 6.6-3). On physical examination the Chvostek sign (Figure 6.6-3) and Trousseau sign (Figure 6.6-4) can be somewhat helpful, but they are not sensitive or specific enough for diagnosis and are mostly of historical interest.
2. Target organ damage: In patients with chronic hypoparathyroidism prolonged hypercalciuria and hyperphosphatemia can result in the development of renal complications such as nephrocalcinosis, nephrolithiasis, and renal insufficiency. Renal function and urine calcium losses should be closely monitored with 24-hour urine calcium measurements as well as assessments of renal function. Nephrocalcinosis or nephrolithiasis can be identified using renal imaging such as ultrasonography. Patients with hypoparathyroidism are at a 3 to 4 times increased risk of renal insufficiency.
Patients with hypoparathyroidism are at risk of developing ophthalmologic complications such as cataracts, which occur in ~50% of cases.
Neurologic complications include intracranial calcification of the basal ganglia. This has been associated with elevations in serum phosphate levels and was noted to occur in the presence of a normal calcium phosphate product. Intracranial calcification has been associated with dystonias and parkinsonism. Brain imaging and electroencephalography are helpful tools in evaluating neurocognitive decline, movement disorders, or seizures in individuals with hypocalcemia.
The standards of care for hypoparathyroidism to prevent target organ damage include:
1) Targeting serum calcium levels (ionized calcium or calcium adjusted for albumin) to mid-low normal reference range in nonpregnant patients.
2) Aiming to target serum phosphate, magnesium, 25-hydroxyvitamin D (25[OH]D) levels within the normal reference range.
3) Avoiding hypercalciuria.
3. Quality of life: Hypoparathyroidism is associated with diminished quality of life. Complaints of “brain fog,” myalgias, numbness, paresthesias, fatigue, weakness, anxiety, and depression all contribute to a lower quality of life. Studies assessing the quality of life in patients with hypoparathyroidism using standardized measurements show that patients have a lower quality of life relative to control populations irrespective of disease duration, etiology, and treatment with activated vitamin D and calcium supplements. Loss of PTH may have significant effects on cognition and overall well-being; this is an area of active research.
DiagnosisTop
For patients with nonsurgical hypoparathyroidism, a detailed family history is required including the presence of consanguinity. Clinical evaluation is used to determine if other features associated with genetic or autoimmune causes of hypoparathyroidism are present. Currently genetic testing is available, enabling a molecular diagnosis of the cause of hypoparathyroidism.
Low serum calcium levels (corrected for low albumin levels if needed) or low ionized calcium levels should be confirmed on ≥2 separate occasions prior to confirming the diagnosis of hypocalcemia. Low PTH levels should also be confirmed on ≥2 occasions at the same time. If PTH levels are not clearly elevated in the presence of hypocalcemia, this confirms the diagnosis of primary hypoparathyroidism, as it indicates an impaired parathyroid response to the low serum calcium levels.
Further biochemical abnormalities that would support the diagnosis of hypoparathyroidism include elevated serum phosphate levels, reductions in calcitriol (1,25-dihydroxycholecalciferol [1,25(OH)2D3]),and elevated urinary calcium excretion.
After parathyroid surgery hypoparathyroidism can be acute and transient (present for <12 months after surgery) or chronic and permanent (present for >12 months after surgery). The diagnosis of permanent surgical hypoparathyroidism is confirmed by the presence of hypocalcemia (ionized calcium or low total serum calcium levels corrected for albumin) for ≥12 months after surgery (in the presence or absence of clinical symptoms) and in the presence of low or inappropriately normal PTH levels on 2 separate occasions.
TreatmentTop
Management requires close monitoring of the biochemical profile and drug therapy to minimize symptoms of hypocalcemia while avoiding overtreatment with the development of hypercalcemia. Prevention of long-term complications requires avoidance of hypercalciuria and hyperphosphatemia. Conventional therapy includes calcium and active vitamin D supplementation. PTH replacement therapy is of value in those in whom conventional therapy has failed.
Acute Hypocalcemia in Hypoparathyroidism
Acute hypocalcemia may require IV calcium, depending on the rate of onset, clinical symptoms, and biochemical severity of hypocalcemia. Symptomatic hypocalcemia requires IV administration of calcium, with the preferred preparation being calcium gluconate. If the total serum calcium level corrected for albumin is <1.75 mmol/L, our advice is that IV calcium should be administered even if the patient is asymptomatic to avoid serious complications of hypocalcemia, including laryngospasm, seizures, or cardiac arrythmias.
A bolus of IV calcium transiently elevates serum calcium for ~2 to 3 hours and a continuous infusion is required to prevent subsequent decreases in serum levels. IV calcium gluconate is preferred to avoid local venous irritation, which can be seen with calcium chloride infusions. Electrocardiographic (ECG) monitoring is required for early detection of any cardiac arrythmias that may occur with calcium infusion.
In the presence of acidosis, the ionized calcium level is higher, as calcium ions are displaced from binding to albumin by hydrogen ions. It is important to correct serum calcium levels prior to correcting acidosis to prevent significant declines in serum calcium concentration.
Avoid infusing sodium bicarbonate or phosphate in the same IV administration line as calcium to prevent precipitation of calcium carbonate or calcium phosphate.
Following the administration of the initial bolus of IV calcium gluconate, a calcium infusion is initiated. A calcium bolus is given over 10 minutes and consists of a 1-g of calcium gluconate in 10 mL of a 10% solution or 90 mg of elemental calcium. Following the bolus, a calcium infusion is started, with 10 ampoules of calcium gluconate containing 900 mg of elemental calcium in 1 L of 5% glucose (dextrose) or normal saline. The infusion is started at 50 mL/h and is titrated to normal serum calcium concentration. To elevate serum calcium levels by 0.5 to 0.75 mmol/L, ~15 mg/kg of elemental calcium IV must be administered over 4 to 6 h.
If the patient is able to take oral calcium supplements, these are also started at the same time as the calcium infusion. Oral calcium salts can be given in the form of calcium carbonate, which contains 40% elemental calcium and is easier to comply with as fewer tablets are required orally. Calcium citrate contains 21% elemental calcium and can be useful in the presence of proton pump inhibitors or achlorhydria, as an acidic pH is not required for absorption of calcium citrate (this is a requirement for the absorption of calcium carbonate). Calcium supplements are initiated in doses of 500 mg to 1000 mg taken with food bid to tid. Patients may require up to 9 g daily. Calcitriol supplementation is of great value as it replaces the deficient 1,25(OH)2D3, which is also low in the absence of PTH. Calcitriol is started at a dose of 0.25 microg once daily or bid and gradually titrated upwards to a maximum of 1 microg daily. Calcitriol has a rapid onset of action with a peak occurring at 3 to 6 hours of administration and a half-life of 4 to 6 hours. If calcitriol is unavailable, alfacalcidol is an alternative, but it is less potent than calcitriol (usual dose, 1-3 microg/d). 25(OH)D stores must also be replenished with cholecalciferol or ergocalciferol and hypomagnesemia (see Hypomagnesemia) must be corrected.
Chronic Hypocalcemia in Hypoparathyroidism
Treatment goals of chronic management are to reduce symptoms of hypocalcemia and the risk of long-term complications of hypercalciuria and hyperphosphatemia. Conventional therapy includes calcium supplementation and active vitamin D and its analogues. Hydrochlorothiazide/thiazide-like diuretics may enhance renal calcium reabsorption and treat hypercalciuria. The data around the benefit of thiazides in patients with hypoparathyroidism are conflicting; clinical judgement must be exercised to assess their utility for patients. Patients started on thiazides must also be cautioned about the increased risk of certain skin cancers associated with thiazide use. Thiazides should not be used in patients with ADH1, since it can exacerbate hypomagnesemia in these patients. They should also be avoided in patients with APS-1 due to its association with adrenal insufficiency; thiazides can worsen hypotension in these patients. Serum magnesium and potassium levels should be closely monitored and corrected as renal losses leading to hypomagnesemia (may cause functional hypoparathyroidism) and hypokalemia are common with thiazide use. 25(OH)D levels should be maintained in the normal reference range of 75 to 125 nmol/L.
Maintaining serum phosphate levels in the normal reference range is important to reduce the risk of extraskeletal calcification as well as the risk of renal, neurologic, and ocular complications associated with hypoparathyroidism. Calcium is an ideal phosphate binder and all patients are advised to take calcium supplements with food, as it binds phosphate in the meal and enables loss of phosphate in the stool. A low-salt diet will also lower urinary calcium losses. There are no data to support the use of other phosphate binders in hypoparathyroidism.
Failure of Conventional Therapy
Failure of conventional therapy is confirmed in the presence of poor control of serum calcium, complications of hypoparathyroidism or its treatment, or poor quality of life; in such cases patients can be offered PTH therapy (see below).
PTH Peptides in the Management of Chronic Hypoparathyroidism
Conventional therapy with calcium and active vitamin D supplementation has been associated with worsening hypercalciuria, renal stones, ectopic calcifications, impaired renal function, and nephrocalcinosis. PTH therapy enhances renal calcium reabsorption and contributes to improved renal phosphate clearance.
PTH therapy can be considered in the presence of:
1) Symptomatic hypocalcemia despite conventional therapy.
2) Requirements of high doses of oral calcium or vitamin D metabolites (>2 g of elemental calcium [calcium carbonate contains ~40% of elemental calcium; calcium citrate, ~20%; and calcium gluconate, 10%] or >1 microg of calcitriol per day).
3) Hypercalciuria, which is defined as a 24-h urinary calcium excretion >6.25 mmol/24 h for adult women and >7.5 mmol/24 h for adult men.
4) Renal insufficiency despite conventional therapy.
5) Hyperphosphatemia.
6) Individuals with poor malabsorption, compliance, or intolerance of large doses of calcium and active vitamin D.
1. PTH(1-34) has a half-life of only 1 hour, and its greatest impact was observed with continuous infusion or multiple daily injections. PTH(1-34) in doses of 20 microg bid resulted in reductions in the dose of calcium and calcitriol required daily and in increased serum calcium while lowering serum phosphate levels.
2. TransCon PTH (palopegteriparitide) is a prodrug consisting of PTH(1-34) linked to polyethylene glycol moiety—a modification of PTH(1-34) that allows to extend its half-life to ~60 hours. In clinical trials the drug has been effective in maintaining eucalcemia while patients are not receiving conventional therapy. It is now approved by the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) as a treatment for hypoparathyroidism.
3. Recombinant human PTH (rhPTH) (1-84; currently unavailable) has been shown to lower urinary calcium losses and serum phosphate levels and allows the patient to decrease the dose of calcium and calcitriol required to maintain target serum calcium concentration. It also makes it possible to achieve normal serum phosphate and urine calcium levels. The majority of patients are able to stop or lower the dose of calcium and calcitriol supplementation required to maintain serum calcium concentration in the low-normal reference range. PTH(1-84) was approved by the FDA to control hypocalcemia in patients with hypoparathyroidism, but due to issues regarding rubber particles in the solution, manufacturing has been stopped and the drug is currently unavailable.
follow-upTop
Recommendations from the standards of care for hypoparathyroidism (the 2022 Canadian and international consensus):
1) Corrected serum calcium levels (albumin-corrected or ionized calcium), phosphorus, magnesium, and creatinine should be monitored every 3 to 6 months. If changes are made in the dose of calcium or calcitriol, or if symptoms of hypocalcemia or hypercalcemia occur, serum calcium and phosphate may require weekly evaluation until stable and in the targeted range.
2) Annual monitoring of urinary calcium excretion, creatinine, and sodium (either by 24-hour urine collection or random urine collection), and 25(OH)D levels is currently advised.
3) Renal imaging by ultrasonography for the presence of nephrocalcinosis or nephrolithiasis is recommended at baseline and is of particular importance in individuals with persistent hypercalciuria, history of renal stones, abnormal urinalysis, or a decline in eGFR.
4) Signs and symptoms of hypercalcemia and hypocalcemia should be evaluated every 6 months, depending on the stability of hypoparathyroidism.
5) The general health of the patient should also be assessed, as many symptoms of hypoparathyroidism are nonspecific and include brain fog and low energy.
6) Basal ganglia calcification can be assessed with brain computed tomography (CT) or susceptibility-weighted magnetic resonance imaging (MRI).
7) An eye examination is of value to determine if cataracts are present.
8) Bone mineral density (BMD) is usually higher than expected for age and gender as evaluated by dual x-ray absorptiometry (DXA). Spinal radiographs may be completed to exclude of vertebral fractures in the presence of significant height loss, back pain, or spinal deformity. Currently there are very limited data on the effects of hypoparathyroidism on fracture risk.
Tables and FiguresTop
|
Postsurgical (75%-80%) |
|
|
Autoimmune (20%-25%) |
Autoimmune polyglandular syndrome-1 |
|
Isolated autoimmune hypoparathyroidism |
|
|
Genetic |
Syndromic: DiGeorge syndrome, CHARGE |
|
Nonsyndromic: Autosomal dominant hypocalcemia type 1 |
|
|
Infiltrative |
Granulomatous: Amyloidosis, sarcoidosis |
|
Metastatic cancer |
|
|
Hemochromatosis |
|
|
Wilson disease |
|
|
Radiation |
|
|
Burn injury |
|
|
Functional hypoparathyroidism |
Hypermagnesemia |
|
Hypomagnesemia |
|
|
Maternal hyperparathyroidism |
|
|
Idiopathic hyperparathyroidism |
|
|
Low serum magnesium |
|
|
Decreased intake or absorption |
– Decreased intake – Malabsorption (short bowel syndrome, steatorrhea, diarrhea, vomiting) |
|
Drugs |
– Diuretics (especially thiazides) – Proton pump inhibitors – Foscarnet, amphotericin B, aminoglycosides, pentamidine, rapamycin – Anticancer drugs (cisplatin, carboplatin) – Immunosuppressants (calcineurin inhibitors: tacrolimus, cyclosporine A) – EGFR inhibitors (cetuximab) |
|
Rare genetic disorders |
– Familial hypomagnesemia with secondary hypocalcemia (TRPM6 gene mutation) – Autosomal dominant hypocalcemia (activating mutation in the CASR gene) – Familial hypomagnesemia with hypercalciuria and nephrocalcinosis |
|
High serum magnesium |
|
|
– Magnesium administration for eclampsia or preeclampsia – Intake in laxatives or cathartics – Metabolic syndromes (familial hypocalciuric hypercalcemia) – Chronic kidney disease |
|
|
EGFR, epidermal growth factor receptor. |
|
| Cause | Symptoms |
|
Hypocalcemia |
– Numbness, tingling in face, hands and feet – Muscle spasms or cramps, laryngospasm – Bradyarrhythmia, heart failure – Wheezing – Depression, confusion, seizures |
|
Hypercalcemia |
– Nausea, vomiting, anorexia – Polyuria, polydipsia – Constipation – Weakness – Confusion – Headaches |

Figure 6.6-3. Chvostek sign is abnormal contracture of the ipsilateral facial muscles that can occur in patients with hypocalcemia. The test is performed by gently tapping over the facial nerve region just anterior to the ear, at the zygomatic arch (~2 cm in front of the earlobe). Twitching of the facial muscles on the same side as the tapping indicates a positive test result. Illustration courtesy of Dr Shannon Zhang.
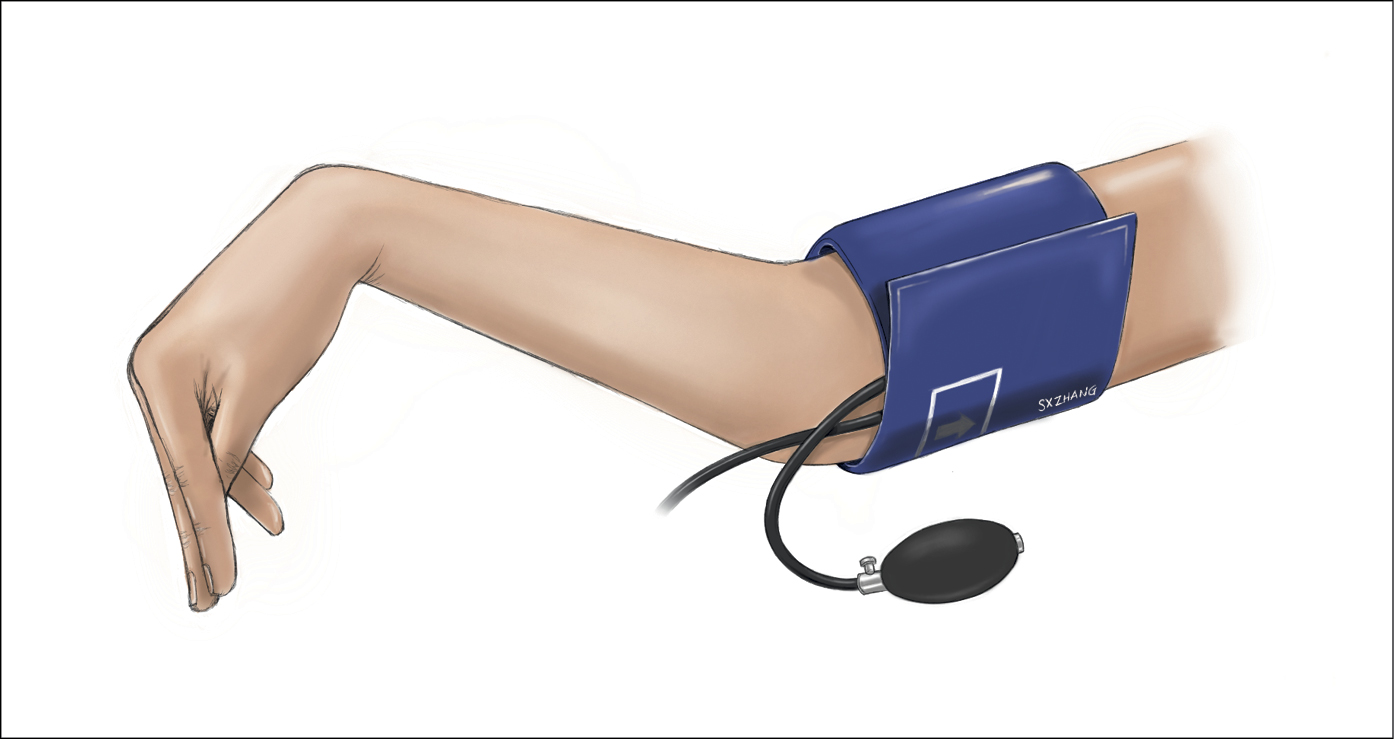
Figure 6.6-4. Trousseau sign is the involuntary carpal spasm (flexion of the wrist, hyperextension of the fingers, and adduction of the thumb) that occurs in a patient with hypocalcemia. The test is performed by inflating a blood pressure cuff 20 mm Hg above the patient’s systolic blood pressure and maintaining the pressure for ≥3 minutes. A positive test result occurs if signs of carpal spasm are noted during this time. Illustration courtesy of Dr Shannon Zhang.