 English
English
 Español
Español
 українська
українська

Ommen SR, Ho CY, Asif IM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines [published correction appears in Circulation. 2024 Aug 20;150(8):e198. doi: 10.1161/CIR.0000000000001277.]. Circulation. 2024;149(23):e1239-e1311. doi:10.1161/CIR.0000000000001250
Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022 May 3;79(17):1757-1780. doi: 10.1016/j.jacc.2021.12.011. Epub 2022 Apr 1. PMID: 35379504.
McDonald M, Virani S, Chan M, et al. CCS/CHFS Heart Failure Guidelines Update: Defining a New Pharmacologic Standard of Care for Heart Failure With Reduced Ejection Fraction. Can J Cardiol [Internet]. 2021 Apr;37(4):531-546. doi: 10.1016/j.cjca.2021.01.017. PMID: 33827756.
Bozkurt B, Coats AJ, Tsutsui H, et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J Card Fail. 2021 Mar 1;S1071-9164(21)00050-6. doi: 10.1016/j.cardfail.2021.01.022. PMID: 33663906.
McDonagh TA, Metra M, Adamo M, et al; ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021 Sep 21;42(36):3599-3726. doi: 10.1093/eurheartj/ehab368. Erratum in: Eur Heart J. 2021 Dec 21;42(48):4901. PMID: 34447992.
Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020 Dec 22;76(25):e159-e240. doi: 10.1016/j.jacc.2020.08.045. Epub 2020 Nov 20. PMID: 33229116.
Bozkurt B, Colvin M, Cook J, et al; American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; and Council on Quality of Care and Outcomes Research. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation. 2016 Dec 6;134(23):e579-e646. Epub 2016 Nov 3. Erratum in: Circulation. 2016 Dec 6;134(23):e652. PMID: 27832612.
WRITING COMMITTEE MEMBERS, Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA Focused Update on New Pharmacological Therapy for Heart Failure: An Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016 Sep 27;134(13):e282-93. doi: 10.1161/CIR.0000000000000435. Epub 2016 May 20. Erratum in: Circulation. 2016 Sep 27;134(13):e298. PMID: 27208050.
Ponikowski P, Voors AA, Anker SD, et al; Authors/Task Force Members. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016 Jul 14;37(27):2129-200. doi: 10.1093/eurheartj/ehw128. Epub 2016 May 20. Erratum in: Eur Heart J. 2016 Dec 30. PMID: 27206819.
Definition, Etiology, PathogenesisTop
Hypertrophic cardiomyopathy (HCM) is a disease of the myocardium that is characterized by an increased left ventricular (LV) wall thickness (>15 mm in any myocardial segment) not explained by conditions that augment LV afterload. Smaller septal thickness (13-14 mm) can be diagnostic in family members of a patient with HCM or in conjunction with a positive genetic test. HCM is an autosomal dominant disease typically due to genetic mutation or mutations of the sarcomeric proteins causing myocardial disarray.
Nearly any pattern of LV wall thickening can be observed in HCM. The most common forms:
1) Asymmetric hypertrophy of the interventricular septum, defined as a septal to posterior wall thickness ratio >1.3 in normotensive individuals or >1.5 in hypertensive individuals. This may result in systolic anterior motion (SAM) of the mitral valve and dynamic obstruction of the left ventricular outflow tract (LVOT).
2) Concentric (symmetric) hypertrophy, defined as an increase in LV wall thickness of all walls not explained by LV loading conditions or infiltrative disease.
3) Midventricular hypertrophy, which can give a “dumbbell” appearance to the LV and cause midcavitary obstruction, sometimes resulting in LV apical dilatation and aneurysm.
4) Apical hypertrophy, defined as apical wall thickness >15 mm or an apical to basal wall thickness ratio >1.3, can give a “spade” appearance to the LV.
Clinical Features and Natural HistoryTop
1. Symptoms: Exertional dyspnea, angina, palpitations, dizziness, syncope or presyncope (particularly in patients with LVOT obstruction).
2. Signs: In hypertrophic obstructive cardiomyopathy (HOCM) a systolic murmur may be heard over the left sternal border with radiation towards the right upper sternal border and the apex. The murmur can intensify with a decrease in preload (eg, during the Valsalva maneuver; after standing from a sitting, lying, or squatting position; or following the administration of nitroglycerin/amyl nitrite) and soften with an increase in afterload (passive elevation of a lower extremity, sitting, squatting, or clenching both fists).
3. Natural history depends on the type and severity of myocardial hypertrophy, LVOT gradient, and propensity for ventricular arrhythmia. Most patients (60%-70%) have a good prognosis and do not experience complications. Those who do develop complications generally fall into one of 3 pathways: sudden cardiac death/ventricular arrhythmias; progressive symptoms from LVOT obstruction or heart failure (HF) with preserved ejection fraction (HFpEF) or HF with reduced EF (HFrEF); or atrial fibrillation (AF).
4. Risk factors for sudden cardiac death (SCD): Nonsustained ventricular tachycardia, LV myocardial thickness ≥30 mm, family history of SCD at a young age (<50 years), unexplained syncope, LVOT gradient >50 mm Hg, apical aneurysm, and LV ejection fraction (LVEF) <50% (not included in recent guidelines). A calculator for the assessment of the risk of SCD in the next 5 years, the HCM Risk-SCD Calculator, can be found at www.mdcalc.com.
DiagnosisTop
HCM is diagnosed morphologically on the basis of results of echocardiography or magnetic resonance imaging (MRI) showing features of myocardial hypertrophy ≥15 mm in a segment of a nondilated LV that cannot be explained only by an increased cardiac load. Genetic testing can be used to confirm the diagnosis and to assess for the presence of HCM in family members.
1. Electrocardiography (ECG): Nonspecific changes; pathologic Q waves, especially in inferior and lateral leads; left axis deviation; abnormal P waves (indicative of enlargement of the left atrium or both atria); in some patients deep T-wave inversions in V2 to V4 (more suggestive of apical variant); features of LV hypertrophy; ventricular and supraventricular arrhythmias.
2. Holter ECG monitoring is performed to detect ventricular tachycardia and atrial fibrillation (AF). In patients with HCM who are at high risk for AF based on risk factors or a validated risk score, extended ambulatory monitoring is now recommended to screen for AF as part of the initial evaluation and annual follow-up. The CHA2DS2-VASc is one such validated risk score (www.chadsvasc.org).
3. Echocardiography (Figure 3.6-3) reveals myocardial hypertrophy, which may occur in any myocardial segment. It should be noted that genotype-positive individuals, including those experiencing SCD, may not have significant hypertrophy on imaging. In patients with asymmetric septal hypertrophy (ASH), there may be elongation of the mitral leaflets, SAM of the mitral apparatus, and LVOT gradient. HCM is considered obstructive when the peak gradient across the LVOT at rest or with provocation (eg, Valsalva maneuver, amyl nitrite) is >30 mm Hg. While there is typically impairment in systolic longitudinal function (detected using Doppler imaging or strain), the overall LVEF may be preserved. LV diastolic function is typically abnormal.
Echocardiography is recommended not only for an initial assessment of every patient suspected of having HCM but also, along with ECG, as a screening study in relatives of patients with HCM. Stress echocardiography may be performed to assess LVOT gradients in patients who do not have significant obstruction at rest and after the Valsalva maneuver or administration of amyl nitrite.
4. ECG stress testing can be performed in patients with unexplained syncope or with symptoms of heart failure particularly to assess for systolic blood pressure decrease during exercise. Pediatric patients with HCM should undergo stress testing regardless of symptom status to determine functional capacity.
5. Exercise echocardiography can be performed in patients with HCM without LVOT gradient at rest, to assess for exercise-induced LVOT obstruction.
6. MRI is recommended when echocardiography results are equivocal, to exclude mimickers, and to aid in risk stratification for SCD. MRI can evaluate LV size, thickness, degree of myocardial fibrosis, and presence of LV aneurysm.
7. Computed tomography (CT) is recommended when echocardiography results are equivocal in patients with contraindications to MRI.
8. Coronary angiography is recommended in patients with a history of cardiac arrest, with sustained ventricular tachycardia, and in all patients ≥40 years with planned invasive treatment of hypertrophy of the interventricular septum.
9. Genetic testing should be offered to all patients with HCM. First-degree relatives may be offered genetic testing if a disease-causing sequence variation is identified in the proband.
Screening of asymptomatic first-degree relatives of patients with HCM should be undertaken with clinical assessment, ECG, and 2-dimensional echocardiography. Screening schedule: Table 3.6-1.
HCM should be differentiated from hypertensive heart disease and athletic heart. Differential diagnosis includes also infiltrative cardiomyopathies (eg, amyloidosis), iron overload, metabolic disorders (eg, Fabry disease, mitochondrial cytopathies, glycogen or lysosomal storage diseases), neuromuscular disease (eg, Friedreich ataxia), or syndromic disorders (eg, Noonan syndrome, LEOPARD syndrome).
TreatmentTop
The basic aims of management are SCD prevention and symptom relief.
Light to moderate exercise is recommended in most patients. Although the risk of SCD may be increased for patients with HCM participating in moderate- to high-intensity competitive sports, it must be assessed individually by providers with expertise in HCM. Avoidance of the Valsalva maneuver and dehydration are generally recommended. There is no specific recommendation against isometric exercise (lack of evidence).
1. Asymptomatic patients: All asymptomatic patients should be assessed for the risk of SCD as this can be the first manifestation of the disease. Pharmacotherapy is not indicated for asymptomatic individuals.
2. Symptomatic patients with LVOT obstruction: Cardioselective beta-blockers titrated to maximum tolerated doses. If beta-blockers are not tolerated, use non-dihydropyridine calcium channel blockers (verapamil, diltiazem) titrated up to a maximum tolerated dose. If symptoms persist with beta-blockers or verapamil, consider adding a myosin inhibitor such as mavacamten (adult patients only; use and titrate in specialized settings) or disopyramide at the maximum tolerated dose in patients with LVOT or intracavity gradients (while monitoring the QTc interval).Evidence 1Weak recommendation (benefits likely outweigh downsides, but the balance is close or uncertain; an alternative course of action may be better for some patients). Low Quality of Evidence (low confidence that we know true effects of the intervention). Quality of Evidence lowered due to the observational nature of data. Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circ Heart Fail. 2013 Jul;6(4):694-702. doi: 10.1161/CIRCHEARTFAILURE.112.000122. Epub 2013 May 23. PubMed PMID: 23704138. Disopyramide should be used in combination with an atrioventricular (AV) nodal blocking agent. Do not use vasodilators (including nitrates and phosphodiesterase inhibitors) and digitalis in patients with LVOT obstruction. Mavacamten has been found to reduce the LVOT gradient and may be used instead of disopyramide. Mavacamten should be discontinued if the patient develops persistent systolic dysfunction (LVEF <50%). It is contraindicated in pregnancy.
3. Symptomatic patients with LVEF >50% without LVOT obstruction: Consider a beta-blocker, verapamil, or diltiazem, and diuretics as needed. For younger patients (≤45 years) with a pathogenic or likely pathogenic genetic variant and mild symptoms valsartan may be beneficial to slow adverse cardiac remodeling.
4. Patients with heart failure and LVEF <50%: Standard therapies for HFrEF should be used, although not specifically studied in this population, including angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), or angiotensin receptor–neprilysin inhibitors (ARNIs), beta-blockers, mineralocorticoid receptor antagonists (MRAs), and sodium-glucose cotransporter-2 (SGLT2) inhibitors.
5. Patients with concomitant AF: This may be poorly tolerated, thus restoration and maintenance of normal sinus rhythm using electrical or chemical cardioversion may be necessary. Anticoagulation is recommended independently of the CHA2DS2-VASc score in those with AF lasting >24 hours. Management of anticoagulation in these patients follows the usual principles, with direct-acting oral anticoagulants being the first-line option. Vitamin K antagonists are the second-line option.
6. Acutely hypotensive patients with LVOT obstruction: If fluid resuscitation is ineffective, IV phenylephrine (or other vasoconstrictors without inotropic activity), alone or in combination with beta-blockers, is recommended. Avoid inotropic agents such as epinephrine, dobutamine, and milrinone.
1. Septal myectomy: First-line therapy to relieve LVOT obstruction in patients with severe symptoms (New York Heart Association [NYHA] class III) and a resting or postexercise peak LVOT gradient ≥50 mm Hg despite receiving maximal tolerated medical therapy.
2. Percutaneous alcohol septal ablation: This is performed by injecting absolute alcohol into a septal perforating arterial branch to induce infarction in a portion of the interventricular septum. Indications are the same as for septal myectomy in patients with contraindications to surgery or those at high surgical risk.
3. ICD implantation is recommended in patients at high risk of SCD (see Clinical Features and Natural History, above) and in patients who have survived cardiac arrest or have recurrent sustained ventricular tachycardia.Evidence 2Strong recommendation (benefits clearly outweigh downsides; right action for all or almost all patients). Moderate Quality of Evidence (moderate confidence that we know true effects of the intervention). Quality of Evidence lowered due to observational data but increased because of the effect size. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000 Feb 10;342(6):365-73. PubMed PMID: 10666426.
4. Heart transplantation may be indicated in patients with end-stage HF or intractable ventricular arrhythmias.
Follow-UpTop
1. In stable patients perform ECG, echocardiography, and 48-hour Holter monitoring every 12 to 24 months. Consider a symptom-limited ECG or stress echocardiography yearly or every 2 to 3 years.
2. In patients with progressive symptoms, repeat echocardiography is suggested.
3. In asymptomatic patients with a gene mutation, perform evaluation (including ECG and transthoracic echocardiography [TTE]) every 2 to 5 years in adults and every 1 to 2 years in children.
Tables and FiguresTop
|
Age of the relative |
Initiation of screening |
Repeat screening |
|
Child and adolescent |
At the time HCM is diagnosed in a family member, or no later than in puberty |
– 1-2 years if early onset disease in family – 2-3 years in most cases |
|
Adult |
At the time HCM is diagnosed in a family member |
Every 3-5 years |
|
HCM, hypertrophic cardiomyopathy. |
||
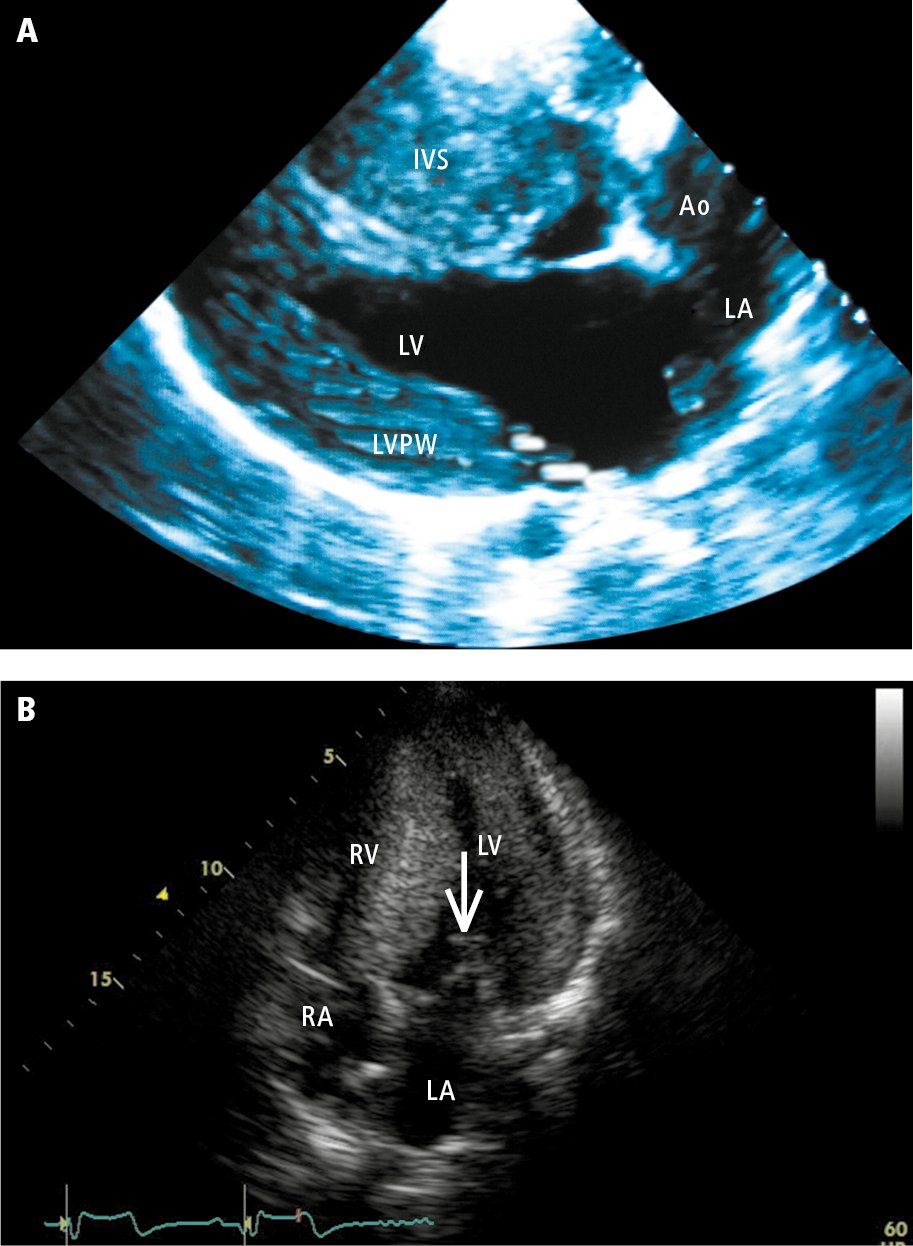
Figure 3.6-3. Echocardiography of patients with hypertrophic cardiomyopathy: A (parasternal view), hypertrophy of the left ventricular posterior wall (PW) and interventricular septum (IVS); B (apical 4-chamber view), systolic anterior motion (SAM) of the anterior mitral leaflet (arrow)—displacement of the anterior leaflet during systole towards the IVS, which results in the obstruction of the left ventricular outflow tract. Ao, aorta; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. Panel B courtesy of Dr Beata Kuśmierczyk-Droszcz.