 English
English
 Español
Español
 українська
українська

Bozkurt B, Colvin M, Cook J, et al; American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; and Council on Quality of Care and Outcomes Research. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation. 2016 Dec 6;134(23):e579-e646. Epub 2016 Nov 3. Erratum in: Circulation. 2016 Dec 6;134(23):e652. PubMed PMID: 27832612.
WRITING COMMITTEE MEMBERS, Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA Focused Update on New Pharmacological Therapy for Heart Failure: An Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016 Sep 27;134(13):e282-93. doi: 10.1161/CIR.0000000000000435. Epub 2016 May 20. Erratum in: Circulation. 2016 Sep 27;134(13):e298. PubMed PMID: 27208050.
Ponikowski P, Voors AA, Anker SD, et al; Authors/Task Force Members. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016 Jul 14;37(27):2129-200. doi: 10.1093/eurheartj/ehw128. Epub 2016 May 20. Erratum in: Eur Heart J. 2016 Dec 30. PubMed PMID: 27206819.
Hilfiker-Kleiner D, Haghikia A, Nonhoff J, Bauersachs J. Peripartum cardiomyopathy: current management and future perspectives. Eur Heart J. 2015 May 7;36(18):1090-7. doi: 10.1093/eurheartj/ehv009. Epub 2015 Jan 29. Review. PubMed PMID: 25636745; PubMed Central PMCID: PMC4422973.
Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014 Oct 14;35(39):2733-79. doi: 10.1093/eurheartj/ehu284. Epub 2014 Aug 29. PubMed PMID: 25173338.
Rapezzi C, Arbustini E, Caforio AL, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013 May;34(19):1448-58. doi: 10.1093/eurheartj/ehs397. Epub 2012 Dec 4. PubMed PMID: 23211230.
Gersh BJ, Maron BJ, Bonow RO, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011 Dec 13;124(24):2761-96. doi: 10.1161/CIR.0b013e318223e230. Epub 2011 Nov 8. PubMed PMID: 22068435.
Charron P, Arad M, Arbustini E, et al; European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010 Nov;31(22):2715-26. doi: 10.1093/eurheartj/ehq271. Epub 2010 Sep 7. PubMed PMID: 20823110.
Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008 Jan;29(2):270-6. Epub 2007 Oct 4. PubMed PMID: 17916581.
Maron BJ, Towbin JA, Thiene G, et al; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006 Apr 11;113(14):1807-16. Epub 2006 Mar 27. PubMed PMID: 16567565.
Definition, Etiology, PathogenesisTop
Restrictive cardiomyopathy (RCM) is a disease of the myocardium characterized mainly by left ventricular (LV) diastolic dysfunction. The causes of RCM are heterogeneous and can be divided into primary (idiopathic RCM and endomyocardial fibrosis) or secondary, many of which are infiltrative (amyloidosis, sarcoidosis, hemochromatosis, systemic sclerosis, carcinoid heart disease, storage diseases, and radiation-induced cardiomyopathy).
Clinical Features and Natural HistoryTop
Manifestations of RCM include dyspnea, fatigue, and features of right heart failure. Arrhythmias are common. The natural history of RCM largely depends on its etiology and severity of myocardial changes.
DiagnosisTop
1. Electrocardiography (ECG) may reveal abnormal P waves, a low R-wave amplitude, flat T waves, and supraventricular arrhythmias, especially atrial fibrillation. In amyloid cardiomyopathy ECG typically has low voltages in limb leads.
2. Echocardiography (Figure 3.6-4) may reveal normal or increased LV wall thickness, enlargement of both atria with relatively small ventricles, normal or slightly impaired systolic function of the ventricles, and diastolic LV dysfunction.
3. Heart catheterization is performed in case of difficulties in differentiating between RCM and constrictive pericarditis.
4. Cardiac magnetic resonance (CMR) imaging may be useful in differentiating underlying etiologies such as sarcoidosis, amyloidosis, endomyocardial fibrosis, or eosinophilia.
5. Cardiac 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) may be useful in conjunction with CMR in sarcoidosis, as it can identify active inflammation and inform on response to therapy.
6. Nuclear scintigraphy with technetium-99m pyrophosphate (99mTc-PYP) can be used to detect cardiac amyloidosis and has the added benefit of distinguishing between the two most common types (immunoglobulin light chain amyloid [AL] and amyloid transthyretin [ATTR]).
7. Endomyocardial biopsy (EMB) is sometimes performed in the setting of suspected infiltrative disease, but the diagnostic yield is low. Noninvasive methods are preferred. EMB should be reserved for cases of diagnostic uncertainty due to equivocal results of noninvasive testing or negative results that are discordant with the degree of clinical suspicion.
RCM is mostly diagnosed on the basis of imaging studies, but in some cases histologic examination of cardiac biopsy specimens is useful.
RCM should be mainly differentiated from constrictive pericarditis. This usually requires a specialized cardiology assessment and may involve invasive testing. It is also important to exclude hypertrophic cardiomyopathy and severe hypertensive heart disease.
TreatmentTop
1. Symptomatic treatment: As in chronic heart failure (see Chronic Heart Failure).
2. Long-term anticoagulation is used in patients with atrial fibrillation.
3. Heart transplant is used in end-stage heart failure not responding to treatment.
4. Treatment of the underlying condition is used in patients with potentially reversible causes.
Special ConsiderationsTop
1. Cardiomyopathy associated with hemochromatosis may lead to heart failure but frequently causes only minor symptoms. Treatment is with phlebotomy.
2. Cardiomyopathy associated with amyloidosis develops secondary to myocardial deposition of amyloid fibrils, leading to significant wall thickening and causing features of restriction. The clinical presentation includes symptoms of heart failure, orthostatic hypotension, and arrhythmias (both atrial and ventricular). In the case of ATTR there may be a history of carpal tunnel syndrome, spinal stenosis, or both. Echocardiographic features include marked ventricular hypertrophy, diastolic dysfunction with or without LV systolic dysfunction, biatrial enlargement, and reduced global longitudinal strain with an apical sparing pattern. Once cardiac amyloidosis is suspected, diagnosis involves screening for amyloid-associated plasma cell dyscrasias (AL) using serum and urine protein electrophoresis and a serum free light chain assay. If a monoclonal protein is identified, urgent hematology referral is warranted. In the absence of a monoclonal protein, a 99mTc-PYP scan should be performed to assess for ATTR. Chemotherapy is available for patients with AL amyloidosis, whereas ATTR can be directly targeted using the stabilizing agent tafamidis. Current guidelines recommend the use of tafamidis in patients with New York Heart Association (NYHA) class I to III symptoms. Caution is advised with angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, digoxin, beta-blockers, and calcium channel blockers, given their predisposition to cause hypotension in patients with amyloid cardiomyopathy.
FiguresTop
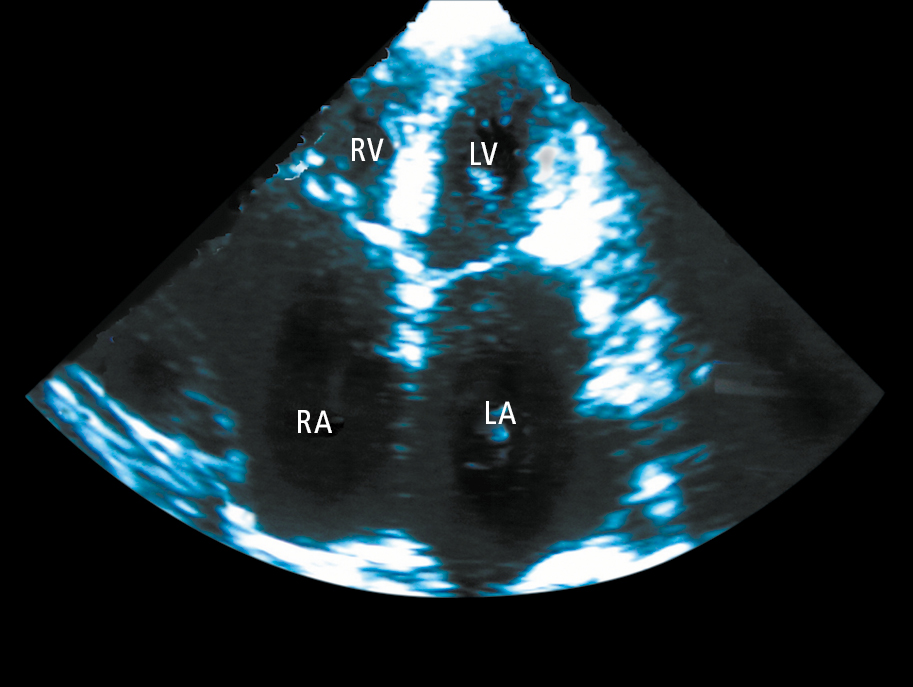
Figure 3.6-4. Echocardiography (apical 4-chamber view) of a patient with restrictive cardiomyopathy showing significant enlargement of both atriums (LA, left atrium; RA, right atrium). The left ventricle (LV) and right ventricle (RV) are normal sized.