 English
English
 Español
Español
 українська
українська

Hirani N, Brunner NW, Kapasi A, Chandy G, Rudski L, Paterson I, Langleben D, Mehta S, Mielniczuk L; CCS/CTS Pulmonary Hypertension Committee. Canadian Cardiovascular Society/Canadian Thoracic Society Position Statement on Pulmonary Hypertension. Can J Cardiol. 2020 Jul;36(7):977-992. doi: 10.1016/j.cjca.2019.11.041. PMID: 32682511.
Helmersen D, Provencher S, Hirsch AM, et al. Diagnosis of chronic thromboembolic pulmonary hypertension: A Canadian Thoracic Society clinical practice guideline update. Can J Resp Crit Care and Sleep Med. 2019.3:4, 177-198, doi: 10.1080/24745332.2019.1631663.
Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019 Jan 24;53(1). pii: 1801913. doi: 10.1183/13993003.01913-2018. Print 2019 Jan. PubMed PMID: 30545968; PubMed Central PMCID: PMC6351336.
Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019 Jan 24;53(1). pii: 1801904. doi: 10.1183/13993003.01904-2018. Print 2019 Jan. PubMed PMID: 30545972; PubMed Central PMCID: PMC6351333.
Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019 Jan 24;53(1). pii: 1801889. doi: 10.1183/13993003.01889-2018. Print 2019 Jan. PubMed PMID: 30545971; PubMed Central PMCID: PMC6351343.
Kim NH, Delcroix M, Jais X, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019 Jan 24;53(1). pii: 1801915. doi: 10.1183/13993003.01915-2018. Print 2019 Jan. PubMed PMID: 30545969; PubMed Central PMCID: PMC6351341.
Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1;37(1):67-119. doi: 10.1093/eurheartj/ehv317. Epub 2015 Aug 29. PubMed PMID: 26320113.
Definition, Etiology, PathogenesisTop
Pulmonary hypertension (PH) refers to an abnormal elevation in mean pulmonary artery pressure (mPAP) at the time of diagnostic right heart catheterization. A recent international task force has proposed a change in the definition from 25 mm Hg to 20 mm Hg with associated increased pulmonary vascular resistance (PVR). Increased mPAP may develop in the course of diseases of the heart, lungs, or pulmonary vessels.
Classification of PH based on pathology according to the European Society of Cardiology:
1) Group 1: Pulmonary arterial hypertension (PAH):
a) Idiopathic.
b) Heritable.
c) Drug-induced or toxin-induced.
d) Associated with connective tissue disease, HIV infection, portal hypertension, congenital heart disease with left to right shunt, schistosomiasis.
e) PAH long-term responders to calcium channel blockers.
f) Pulmonary veno-occlusive disease (PVOD), pulmonary capillary hemangiomatosis, or both.
g) Persistent PH of the newborn.
2) Group 2: PH due to left heart disease (heart failure with preserved or reduced left ventricular ejection fraction; valvular disease; congenital/acquired cardiovascular condition leading to postcapillary PH).
3) Group 3: PH due to lung diseases, hypoxia, or both (chronic obstructive pulmonary disease [COPD], restrictive lung disease, other pulmonary diseases with mixed restrictive/obstructive pattern, hypoxia without lung disease [severe sleep-disordered breathing including obesity hypoventilation syndrome and exposure to high altitude], developmental lung disorders).
4) Group 4: Chronic thromboembolic PH (CTEPH) and other pulmonary artery obstructions.
5) Group 5: PH with unclear or multifactorial etiology (or both):
a) Hematologic disorders (chronic hemolytic anemia, myeloproliferative neoplasms, splenectomy).
b) Systemic disorders (sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis).
c) Metabolic disorders (glycogen storage diseases, Gaucher disease, thyroid disorders).
d) Other: Pulmonary tumoral thrombotic microangiopathy, fibrosing mediastinitis, chronic renal failure treated with or without hemodialysis, segmental pulmonary hypertension.
Clinical Features and Natural HistoryTop
1. Symptoms of isolated PH: Progressive limitation of exercise tolerance caused by dyspnea (the key symptom, regardless of etiology). Initial symptoms are mild and nonspecific. Resting dyspnea is often absent even in patients with advanced PH. Angina may be present, which is caused by right ventricular ischemia or by compression of the left main coronary artery by a significantly dilated pulmonary artery. Compression of the recurrent laryngeal nerve by dilated pulmonary arteries may cause hoarseness. Symptoms of the underlying condition are also seen (eg, heart failure or systemic disease, especially scleroderma). The World Health Organization (WHO) functional classification is determined on the basis of symptom severity (Table 1).
2. Signs: Systolic parasternal heave, increased pulmonary component of the second heart sound, and murmur of tricuspid regurgitation. Features of right ventricular heart failure (see Chronic Heart Failure) and of the underlying condition.
3. Natural history: The disease is usually progressive, especially in patients with PAH (group 1).
DiagnosisTop
Diagnosis includes confirmation of PH and determination of its cause.
1. Laboratory investigations: There are no laboratory features specific for isolated PH. The following tests may sometimes yield abnormal results, which are usually due to diseases associated with PH:
1) Arterial blood gas analysis may reveal moderate hypoxemia (this may be significant in patients with interstitial lung disease, congenital heart disease with shunt reversal, and in patients with a right-to-left shunt through a patent foramen ovale) or hypercapnia in patients with COPD or central abnormalities of respiratory function.
2) Antinuclear antibodies are found in approximately a third of patients with idiopathic PH. Anticentromere antibodies in the setting of scleroderma are particularly relevant.
3) HIV serology should be routinely measured.
4) Abnormal liver function tests are found in patients with PH associated with portal hypertension (portopulmonary hypertension).
2. Electrocardiography (ECG) (Figure 1) is often normal in patients with early PH. In more advanced disease ECG reveals right axis deviation, P pulmonale, right bundle branch block, right ventricular hypertrophy, and right ventricular overload. Patients may also have arrhythmia (most frequently atrial tachycardia and atrial flutter).
3. Chest radiography (Figure 2) reveals dilation of the main pulmonary artery, lobar pulmonary arteries, right ventricle, and right atrium. Pulmonary congestion is seen in patients with PH due to left ventricular (LV) failure and in PVOD (without LV dysfunction documented on echocardiography). Features of interstitial lung disease or emphysema may also be seen.
4. Pulmonary function tests:
1) Spirometry: Results are often normal. Features of pulmonary restriction or obstruction are seen in patients with PH caused by interstitial lung disease or airway disease, respectively. Mild obstruction of the small airways may also be present.
2) Plethysmography: In the presence of restriction, interstitial lung disease should be considered and correlated with evidence of pulmonary fibrosis on high-resolution computed tomography (HRCT).
3) Carbon monoxide diffusing capacity (DLCO): This may be decreased in patients with idiopathic PH and is particularly low in patients with concomitant parenchymal lung disease.
5. Echocardiography (Figure 3): The primary screening test used in the evaluation of PH. It has the advantage of identifying the etiology responsible for PH by evaluating for LV systolic and diastolic dysfunction, valvular heart disease, congenital heart disease, and other structural abnormalities. Features associated with PH include right ventricular dilation and right ventricular hypokinesis; in more advanced disease, right atrial enlargement, decreased dimensions and deformation of the LV and left atrium, and presence of pericardial effusion are also apparent. Doppler echocardiography, particularly when focused on the assessment of regurgitant jets through the right-heart valves, is used to estimate the pulmonary artery pressures.
6. Perfusion scintigraphy (Figure 4) is critical in the evaluation of PH and should be considered in most patients. In the setting of CTEPH, ventilation-perfusion (V/Q) scans have exceptional sensitivity and a negative predictive value. As such, the use of V/Q imaging to exclude CTEPH is essential, given the dramatically different therapeutic approach and prognostic implications of CTEPH in comparison with other forms of PH. Identified perfusion defects should be correlated with computed tomography pulmonary angiogram (CTPA) to confirm the presence of intravascular filling defects.
7. Chest computed tomography (CT) demonstrating main pulmonary artery enlargement (≥29 mm), right ventricular dilation, right atrial dilation, or a main pulmonary artery to ascending aorta diameter ratio ≥1 is suggestive of PH. Unenhanced cuts can identify parenchymal lung disease and discriminate between group 3 and group 1 PH. Characteristic findings of PVOD can also be observed on CT. Contrast enhancement of the pulmonary vasculature is critical for the evaluation of CTEPH.
8. Catheterization of the right heart and main pulmonary artery is the gold standard in the hemodynamic assessment of pulmonary circulation and should be completed prior to the initiation of PAH-targeted therapies to provide critical diagnostic and prognostic information. In patients with idiopathic, heritable, or drug-induced PAH, perform acute vasoreactivity testing using a potent pulmonary artery vasodilator (nitric oxide, prostacyclin, or IV adenosine) to assess for vasodilator response, the presence of which has critical treatment and prognostic implications. Hemodynamic evaluation should be completed at dedicated centers of excellence with experience in managing patients with PAH.
9. Other studies depend on the suspected underlying condition.
The probability of PH is assessed using noninvasive techniques. Echocardiography is the primary screening test used in patients suspected to have PH. The probability of PH can be determined using the peak tricuspid regurgitation velocity to estimate right pulmonary artery systolic pressure and a formal evaluation of the size and function of the right-sided cardiac chambers as outlined below:
1) ≤2.8 m/s (tricuspid valve pressure gradient [TVPG] ≤31 mm Hg): Low probability.
2) 2.9 to 3.4 m/s (TVPG, 32-46 mm Hg): Intermediate probability.
3) >3.4 m/s: High probability.
The probability is increased (from low to intermediate and from intermediate to high) in the presence of other echocardiographic features of right ventricular or atrial overload (eg, a right ventricle to LV basal diameter ratio >1.0, flattening of the interventricular septum, pulmonary artery diameter >25 mm, inferior vena cava diameter indicating raised pressure). The echocardiographic probability of PH and clinical features are the basis for indications for cardiac catheterization, which is required to secure a formal diagnosis of PH.
In patients with no clear evidence suggesting a definite PH etiology that would qualify them to group 2, 3, or 5, the echocardiographic diagnosis should be confirmed by direct pressure and flow measurements during pulmonary artery catheterization (mean pressure at rest ≥20 mm Hg and PVR >3 Wood units). Cardiac catheterization is also useful in the differential diagnosis of PH. A pulmonary capillary wedge pressure >15 mm Hg is the key diagnostic criterion of PH caused by left-sided heart disease. Right atrial pressure, cardiac index, pulmonary vascular resistance, and mixed venous oxygen saturation are strong prognostic factors.
To establish prognosis and treatment, additionally assess the patient’s WHO functional class (Table 1), results of the 6-minute walk test, and serum B-type natriuretic peptide (BNP) levels.
TreatmentTop
Treatment depends on the appropriate clinical classification of PH. Patients with both PAH and CTEPH are managed aggressively, as treatment has the potential to dramatically impact the clinical course and represents a cure for some patients. The treatment regimen for PAH has evolved dramatically over the last 25 years with the availability of targeted PAH therapies. Combination therapy, implemented in a goal-directed manner, has had a marked impact on disease course. Given the potential benefits and toxicities of these therapies, their prescription and management should be orchestrated through an experienced PH center of excellence.
1. PAH: Management should include acute vasoreactivity testing in patients with idiopathic, heritable, or drug-induced disease. In patients with vasoreactivity (~15% of patients with idiopathic PAH) start treatment with a calcium channel blocker. Other PAH patients are managed with PAH-specific therapies that typically should be used in combination to achieve optimal outcome. These agents primarily target endothelial cell dysfunction and include phosphodiesterase-5 (PDE-5) inhibitors, soluble guanylate cyclase stimulators, endothelin receptor antagonists, and/or agents targeting the prostacyclin pathways. Recent European guidelines suggest monotherapy in a minority of patients and only in specific situations.
2. PH due to left heart disease: Management involves treatment and optimization of the underlying cardiac condition. Randomized clinical trials have not shown a clear role for PAH-specific therapies in this patient population.
3. PH due to lung disease: Treatment of the underlying condition, oxygen therapy, consideration of lung transplant.
4. CTEPH: Lifelong anticoagulation. In patients with WHO functional class II to IV, pulmonary endarterectomy is the gold-standard therapeutic intervention. Pulmonary endarterectomy is completed with curative intent with normalization of pulmonary hemodynamics in the majority of patients. In those who do not qualify for pulmonary endarterectomy because of distal surgically inaccessible disease or medical comorbidities, targeted medical therapy with the soluble guanylate cyclase stimulator riociguat with or without balloon pulmonary angioplasty (BPA) has therapeutic value.
1. Patients with PH should be encouraged to be active within symptom limits. They should avoid excessive physical activity that leads to distressing symptoms, especially angina and syncope. Supervised exercise rehabilitation should be considered in deconditioned patients.
2. Dietary salt restriction, avoidance of excessive fluid intake.
3. Avoidance of pregnancy in women of child-bearing age.
4. Immunization against influenza and pneumococcal infection.
5. Psychosocial support through a multidisciplinary care team.
1. Anticoagulant treatment: Oral therapy may be considered in patients with idiopathic PAH, hereditary PAH, or PAH secondary to anorexigens, although the registry and randomized controlled trial data supporting this indication are weak and contradictory. Do not use anticoagulant treatment in patients with Eisenmenger syndrome or connective tissue disease because of the risk of bleeding. PH associated with LV dysfunction or lung disease is not an independent indication for anticoagulant treatment.
2. Oxygen therapy: Continuous supplemental oxygen therapy is recommended when partial pressure of oxygen in arterial blood (PaO2) is ≤60 mm Hg.
3. Diuretics: These are used in case of right ventricular failure (see Table 2 in Chronic Heart Failure).
4. Calcium channel blockers: Treatment exclusive to patients with documented hemodynamic evidence of pulmonary vasoreactivity (see Diagnostic Tests, above). Use nifedipine up to 240 mg/d, diltiazem up to 720 mg/d, or amlodipine up to 20 mg/d (agents: see Table 4 in Essential Hypertension). Start from a standard dose and titrate upwards based on the patient’s treatment tolerance.
5. “Targeted” treatment recommended in group 1 PAH: International guidelines recommend initiation of such therapies under the supervision of an expert treatment center. Agents are often used in combination in a goal-oriented fashion. Parenteral therapies are reserved for patients with the most advanced disease.
1) PDE-5 inhibitors: Sildenafil 20 mg tid, tadalafil 40 mg once daily (these are used in PAH). Common adverse effects include headache, flushing, dyspepsia, and visual disturbance. Concurrent use of organic nitrates in any form is contraindicated.
2) Endothelin receptor antagonists: Bosentan 62.5 to 125 mg bid, ambrisentan 5 to 10 mg once daily, macitentan 10 mg once daily. The most common adverse effect of endothelin receptor antagonists is increase in serum aminotransferase (monitor the levels during treatment); the increase is usually transient and asymptomatic, although it may warrant discontinuation of the drug. Ambrisentan and macitentan have lower hepatotoxicity.
3) Prostanoids are used in patients with severe disease as reflected by poor hemodynamic, functional, and clinical parameters. Without such intervention, the long-term prognosis in these patients is poor. Epoprostenol improves symptoms, exercise capacity, and hemodynamics, and is the first treatment shown to reduce mortality. Epoprostenol is administered by continuous IV infusion (using a central venous catheter and infusion pump). Abrupt interruption of epoprostenol infusion should be avoided because in some patients this may lead to a PH rebound with symptomatic deterioration and even death. Treprostinil, a prostacyclin analogue, is administered by continuous subcutaneous infusion using a microinfusion pump and small abdominal subcutaneous catheter, IV, or orally. Iloprost, a prostacyclin analogue, is inhaled (6-9 times a day). Selexipag is a novel prostacyclin receptor agonist that can be administered orally.
4) Riociguat is a stimulator of soluble guanylyl cyclase approved by the US Food and Drug Administration for treatment of group 1 patients and the first-line agent in patients with CTEPH who do not qualify for pulmonary endarterectomy. Doses, ranging from 1 to 2.5 mg tid, are established individually. Do not use riociguat in combination with a PDE-5 inhibitor due to the high risk of hypotension and increased risk of death.
1. Balloon atrial septostomy (BAS) is performed to create a septal defect to offload a failing right ventricle. This is a palliative procedure, sometimes also used as a bridging procedure before lung transplant.
2. Bilateral lung transplant or heart and lung transplant is indicated in patients with unequivocal progression of PH despite using all available medical treatment measures.
3. CTEPH:
1) Pulmonary endarterectomy (treatment of choice in patients with symptomatic CTEPH) is the mechanical removal of thrombi attached to the endothelium of the proximal sections of pulmonary arteries.
2) BPA may be an alternative to surgical treatment mainly in patients who do not qualify for endarterectomy due to excessively distal location of lesions, very advanced age, or serious comorbidities.
PrognosisTop
Historically, the average survival time in patients with idiopathic PAH was 2 to 3 years and in those with functional class IV, <6 months. With the availability of targeted PAH therapies, the natural history of disease has been transformed, resulting in 5-year survival rates of 65%. Despite the availability of several effective therapeutic options, many patients are still at risk of substantial morbidity and premature mortality. In patients with evidence of vasoreactivity, the 5-year survival rate is 95%.
Tables and FiguresTop
|
Class |
Description |
|
I |
Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnea or fatigue, chest pain, or near syncope |
|
II |
Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnea or fatigue, chest pain, or near syncope |
|
III |
Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnea or fatigue, chest pain, or near syncope |
|
IV |
Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. These patients manifest signs of right-heart failure. Dyspnea, fatigue, or both may even be present at rest. Discomfort is increased by any physical activity |
|
Source: Eur Heart J. 2016;37(1):67-119. | |
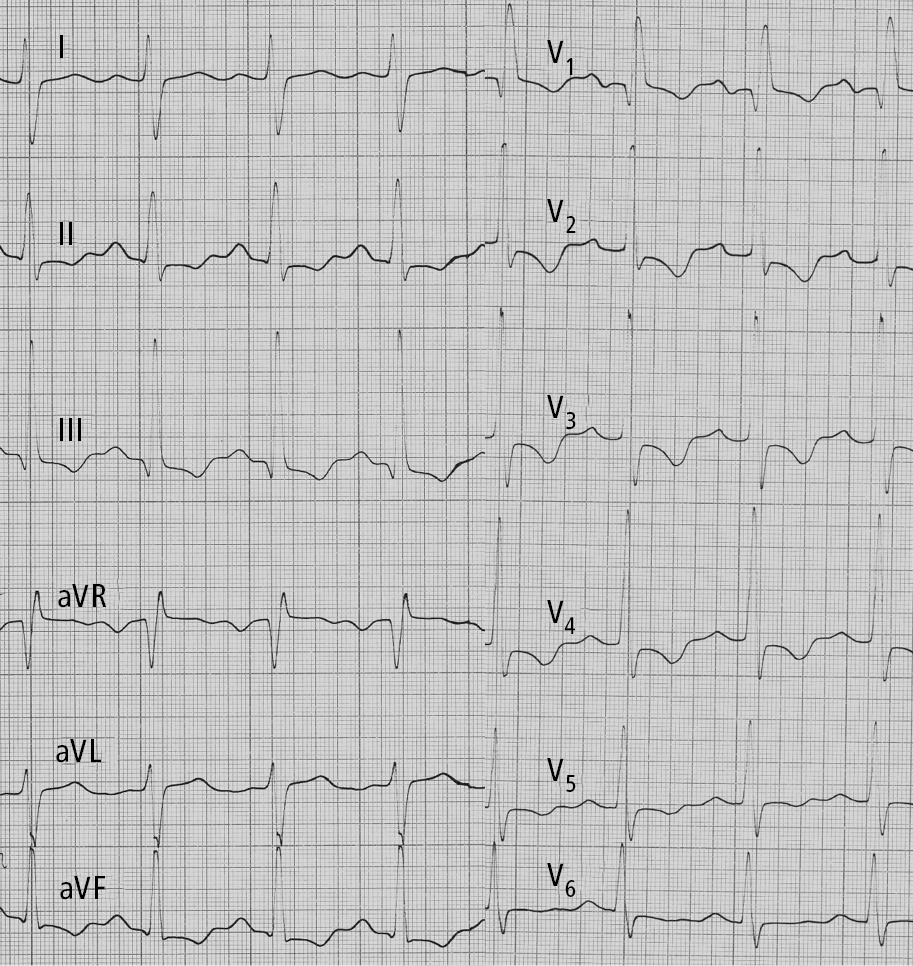
Figure 17.18-1. Electrocardiography (ECG) of a patient with idiopathic pulmonary hypertension: right axis deviation, P pulmonale, right bundle branch block, features of right ventricular hypertrophy and right ventricular strain. Figure courtesy of Dr Adam Torbicki.
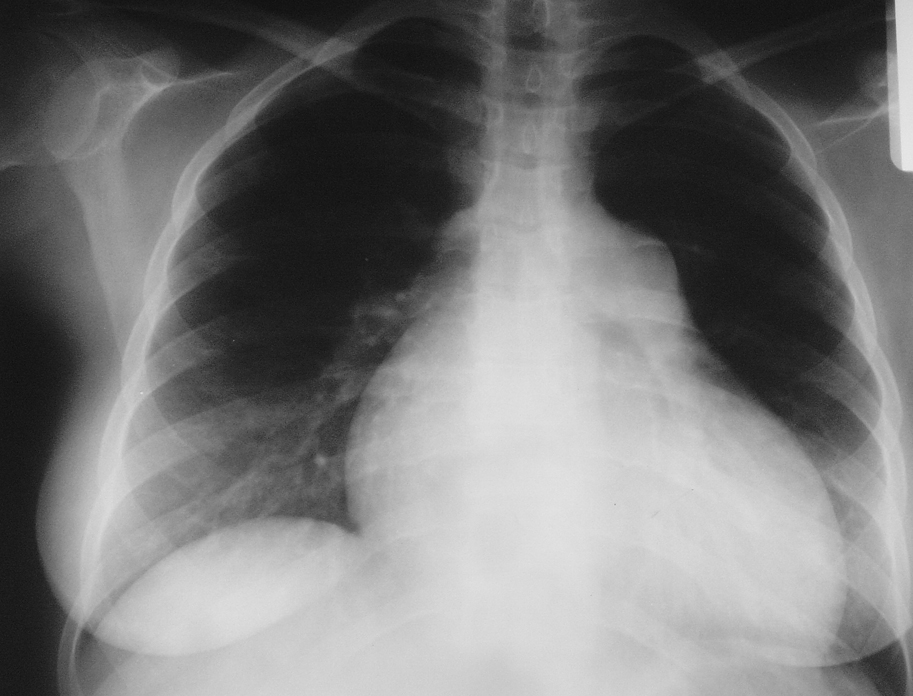
Figure 17.18-2. Chest radiography of a patient with idiopathic pulmonary hypertension: enlargement of the right ventricle and right atrium, prominent main pulmonary artery. No focal lesions in the lungs. Figure courtesy of Dr Adam Torbicki.
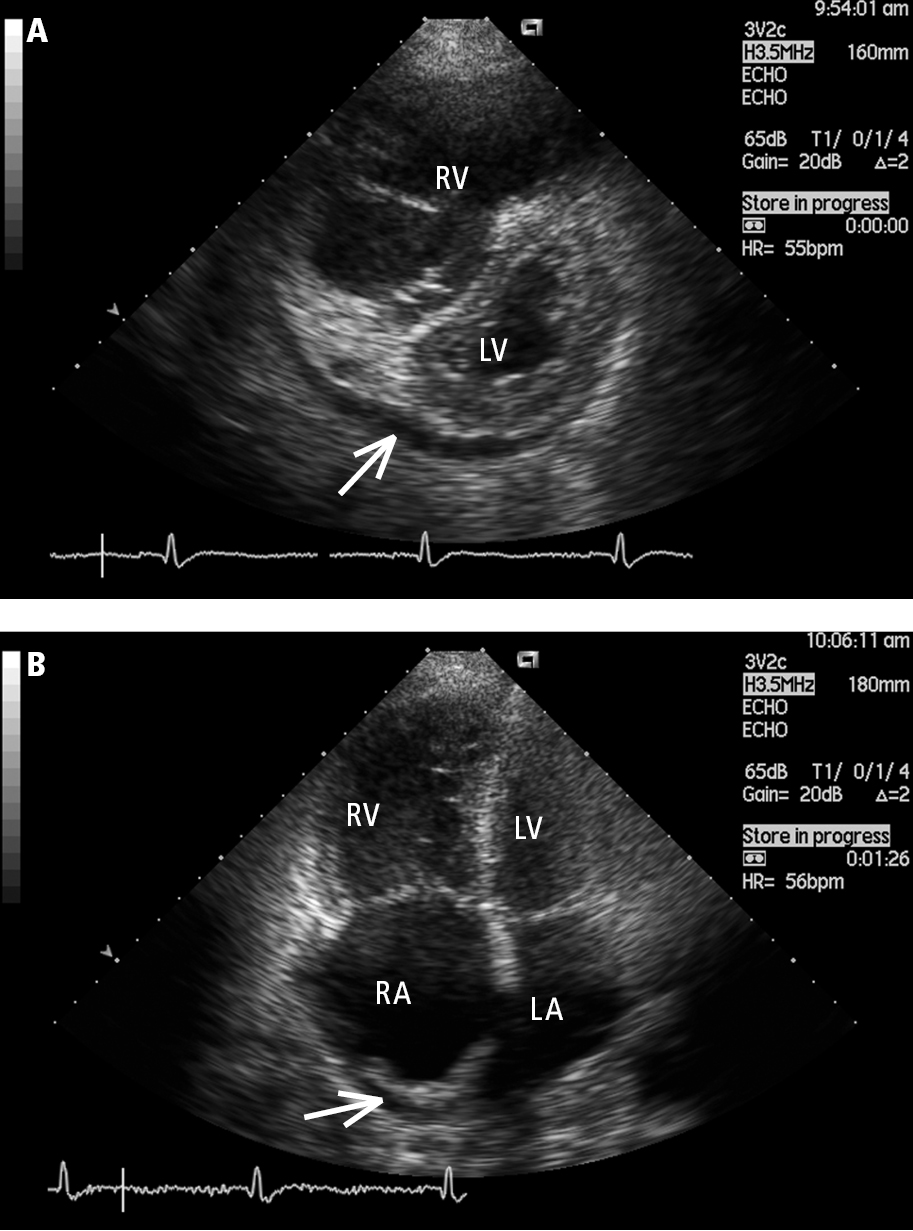
Figure 17.18-3. Transthoracic echocardiography (TTE), parasternal short-axis view (A) and apical 4-chamber view (B): a markedly enlarged right ventricle (RV) in a patient with idiopathic pulmonary hypertension. Left heart compression, right atrial (RA) dilation, and pericardial effusion (arrow) are markers of poor prognosis. LA, left atrium; LV, left ventricle. Figure courtesy of Dr Adam Torbicki.
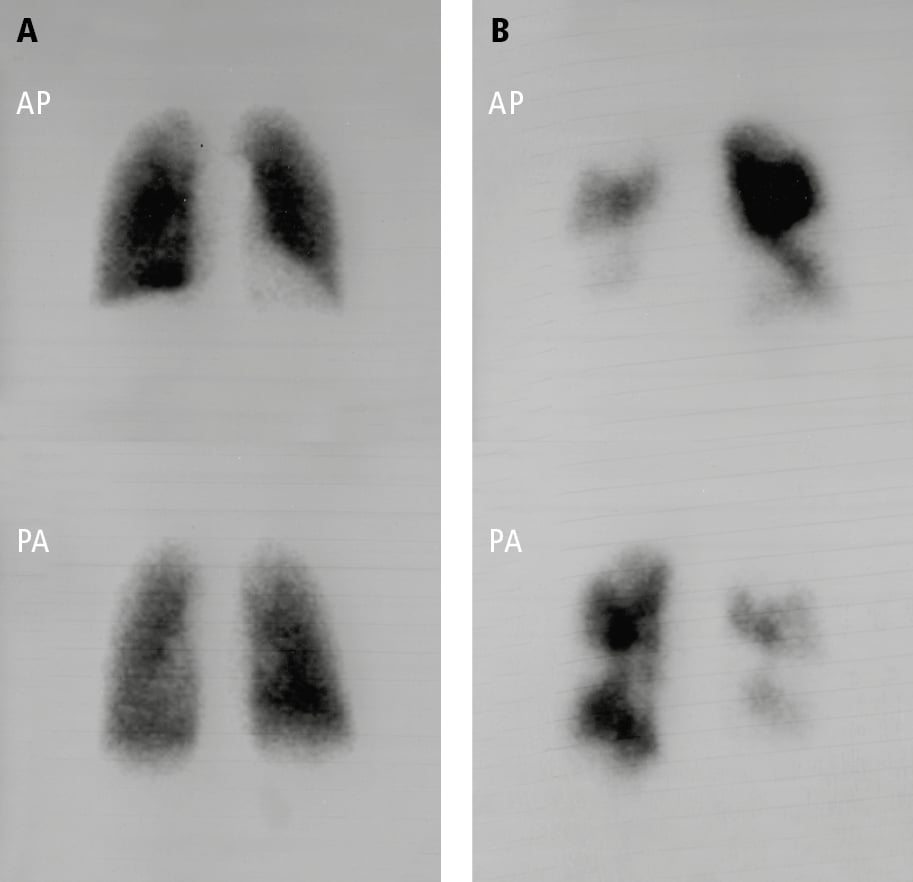
Figure 17.18-4. Pulmonary perfusion scintigraphy: A, normal pulmonary perfusion in a patient with pulmonary arterial hypertension secondary to systemic sclerosis; B, extensive perfusion defects in both lungs in a patient with thromboembolic pulmonary hypertension. AP, anteroposterior view; PA, posteroanterior view. Figure courtesy of Dr Adam Torbicki.