 English
English
 Español
Español
 українська
українська

Bernasconi A, Cendes F, Theodore WH, et al. Recommendations for the use of structural magnetic resonance imaging in the care of patients with epilepsy: A consensus report from the International League Against Epilepsy Neuroimaging Task Force. Epilepsia. 2019 Jun;60(6):1054-1068. doi: 10.1111/epi.15612. Epub 2019 May 28. PMID: 31135062.
Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017 Apr;58(4):522-530. doi: 10.1111/epi.13670. Epub 2017 Mar 8. PMID: 28276060.
Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014 Apr;55(4):475-82. doi: 10.1111/epi.12550. Epub 2014 Apr 14. PMID: 24730690.
Beghi E, Carpio A, Forsgren L, et al. Recommendation for a definition of acute symptomatic seizure. Epilepsia. 2010 Apr;51(4):671-5. doi: 10.1111/j.1528-1167.2009.02285.x. Epub 2009 Sep 3. PMID: 19732133.
Von Oertzen J, Urbach H, Jungbluth S, et al. Standard magnetic resonance imaging is inadequate for patients with refractory focal epilepsy. J Neurol Neurosurg Psychiatry. 2002 Dec;73(6):643-7. doi: 10.1136/jnnp.73.6.643. PMID: 12438463; PMCID: PMC1757366.
Definition, Etiology, PathogenesisTop
An epileptic seizure is a clinical symptom of aberrant electrical discharge within the brain. Detailed seizure symptomatology (ie, semiology) is dependent on the pattern of discharge, including its localization, distribution, intensity, duration, speed, and anatomic pathway of current spread.
Epilepsy is a chronic condition in which epileptic seizures manifest in a recurrent fashion, either unprovoked or provoked in response to triggering factors. It is possible to define the risk of seizure recurrence as early as after a first seizure, meaning that epilepsy can be diagnosed after a single seizure given specific findings on brain imaging, recording of electrical brain activity via electroencephalography (EEG), or both. While the diagnosis of epilepsy requires the occurrence of seizures, not all seizures imply epilepsy, as epileptic seizures can represent a temporary symptom related to acute brain dysfunction (acute symptomatic seizures [ASSs]; also see Acute Seizures).
The etiology of seizures and epilepsy encompasses a large spectrum of central nervous system (CNS) diseases, including various congenital and acquired structural brain lesions (Figure 12.3-1). The main etiologic categories are developmental malformations, vascular malformations, neoplasms, traumatic lesions, (post)inflammatory and (post)infectious lesions, neurovascular disease, and—as a lesion of special significance—hippocampal sclerosis. Genetic anomalies may account for certain epileptogenic CNS lesions (eg, tuberous sclerosis, Sturge-Weber disease, selected CNS malformations) or an underlying propensity for electrical discharge (eg, mutations in genes encoding ion channels). ASSs may be seen in a variety of neurologic conditions, including acute illness and disturbances of homeostasis or as a response to drugs (Table 12.3-1).
On a cellular level, the long-term activity profile of epileptic neurons is altered in a way that produces a more erratic discharge pattern and the neurons no longer fire single action potentials in an orderly way. Instead, epileptic neurons tend to fire in burst activity (paroxysmal depolarization shift), often in groups (hypersynchrony), and they form oscillating networks. Glia and other cells are also involved in epileptogenic dynamics. Because epilepsy often manifests later in life after a CNS insult or in case of a congenital lesion, an occult epileptogenic process evolving over time can be proposed, which currently cannot be targeted therapeutically. Network hyperexcitability in epilepsy not only relates to aberrant excitatory cellular processes but also includes aberrant CNS inhibition.
Clinical Features and Natural HistoryTop
1. Signs and symptoms: Seizures and associated symptoms are classified as focal, generalized, or unknown onset. The terms “focal” and “generalized” refer to the electrical discharge mechanism: focal means the electrical discharge is generated in a rather localized part of one hemisphere, whereas generalized means the discharge pattern at onset is more widespread and usually involves both hemispheres. Focal-onset seizures are further classified clinically according to whether the patient’s awareness is preserved (previously known as “simple partial”) or impaired (previously known as “complex partial”). Both focal and generalized seizures may present with motor and/or nonmotor signs and symptoms.
Classic grand mal or tonic-clonic seizure (TCS): A TCS is a rather stereotyped sequence of bilateral extensor stiffening, often initiated by vocalization (“epileptic cry”) and followed by bilateral symmetric clonic jerking. This type of epileptic fit generally evolves for 1 to 2 minutes and ends with a transient coma that resolves gradually. A TCS can develop as a primary generalized seizure or evolve from a focal seizure (secondary generalized or focal to bilateral tonic-clonic). Of note, many epilepsy patients have no history of TCSs.
1) Types of classic focal seizures:
a) Jacksonian seizure: Unilateral motor (clonic twitching/jerking) or sensory signs (paresthesias) restricted to one part of the body, often evolving to neighboring body parts (the Jacksonian march). This seizure type arises in or next to the central region (precentral and postcentral gyrus).
b) Focal seizure of temporal lobe origin: An episode of impaired awareness (motionless stare, amnesia) often combined with automatisms, such as lip smacking, chewing, or swallowing. It lasts between 30 and 90 seconds and evolves gradually (although it can occur with great variation, in exceptional cases with awareness preserved).
c) Focal seizure of frontal lobe origin: Often associated with bizarre motor behavior, abrupt beginning and ending, and rather brief duration. Impairment of consciousness is variable.
d) Focal aware seizures with subjective symptoms: The focal discharge is causing sensory symptoms (eg, optic, acoustic, olfactory, gustatory, somatosensory, vertiginous) or autonomic changes (eg, epigastric rising sensation, nausea, piloerection, tachycardia). Such symptoms often initiate other seizure forms as auras (ie, a specific aura represents a focal aware seizure).
Any focal seizure can evolve into a generalized seizure, which most often is a TCS (ie, focal to bilateral tonic-clonic seizure; secondary generalized seizure). Focal seizures can be followed by transient specific neurologic deficits, such as localized weakness (Todd paralysis), numbness, aphasia, or visual field impairment; these Todd phenomena resolve mostly within 48 hours and can give important clues as to the affected hemisphere.
2) Types of classic generalized seizures:
a) Primary generalized TCS: Note that the mere absence of focal features does not exclude a focal origin.
b) Absences: An absence is a sudden interruption of the conscious stream lasting seconds. With the abrupt ending, the patient returns to normal.
c) Myoclonic seizures: Rapid brief muscular jerks, often bilateral and symmetric, frequently affecting the upper extremities, and most often occurring with preserved consciousness.
Mild seizure symptoms frequently remain undiagnosed and can occur for years before the first TCS; such seizures can be difficult to recognize if they occur without ever evolving into a TCS. There are also miscellaneous other seizure types, which are beyond the scope of this chapter.
A special manifestation of seizures is status epilepticus, which corresponds to persistent seizure activity that is not self-limiting. Status epilepticus can appear as a convulsive status (tonic-clonic status, focal motor status), nonconvulsive status (different types), or epilepsia partialis continua.
2. Natural history: It is not well understood why brain insults are followed by epilepsy in some patients but not in others. This points to multiple factors that may contribute to seizure generation, including different pathophysiologies and genetic predispositions. The natural history of the clinical course of epilepsy is difficult to delineate, because most epilepsy cases are treated once a diagnosis is made. With adequate pharmacotherapy two-thirds of patients remain seizure free, while one-third continue to have seizures (refractory epilepsy). Different epilepsies have different courses and response rates. A clinical course with only a few seizures is possible in some patients that are not receiving pharmacotherapy, but this scenario is not very common. Some epilepsies are age dependent and may show complete remission over time, especially among certain types (eg, childhood absence epilepsy or benign epilepsies of childhood [eg, rolandic epilepsy]).
DiagnosisTop
Diagnoses of seizures and epilepsy can be made on clinical grounds alone in a high percentage of patients. Therefore, accurate history taking from both the patient and witnesses forms the foundation of a correct diagnosis and should not be undervalued. Ancillary tests do not necessarily yield abnormal results.
At first presentation, the following should be evaluated:
1) Core symptoms that occurred (sequence, evolution).
2) Symptom(s) that constitute the initial phase (aura).
3) Presence of specific postictal deficits (Todd phenomena).
4) Presence of specific triggers (precipitants).
5) Pattern of occurrence (daytime, sleep, linked to the menstrual cycle).
6) Occurrence of different kind of episodes.
7) Risk factors for epilepsy (early life illnesses, family history).
These questions are helpful in collecting the core answers:
1) Is the event a seizure?
2) Is this seizure really the first one?
3) Are there other seizure types?
4) Is it epilepsy?
5) Is it a focal or generalized epilepsy syndrome?
The prevailing goal of epilepsy classification is the dichotomic separation of focal epilepsy versus generalized epilepsy, which follows the rationale of differentiating between more localized and widespread electrical disturbances as the defining pathophysiology. Differentiating between these two categories is the minimum aim in epilepsy care, as the diagnostic, therapeutic, and prognostic implications differ widely. For example, in focal epilepsy it is vital to vigorously search for structural CNS lesions, which are absent in classic generalized epilepsies (idiopathic generalized epilepsies [IGEs]); drug selection is different in IGEs and in focal epilepsies; and focal epilepsies may be treated by epilepsy surgery, which is not an option in IGEs.
An epilepsy syndrome refers to classic combinations of signs and symptoms in epilepsy. Well-established IGE syndromes include childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and generalized epilepsy with grand mal only.
IGEs often present with a TCS, and/or absence, and/or myoclonic seizures in distinct syndromic patterns. Focal epilepsies usually present with milder seizure types in addition to TCSs. A very common focal epilepsy syndrome is mesial temporal lobe epilepsy due to hippocampal sclerosis (MTLE-HS), in which patients develop classic temporal lobe seizures (eg, aura with epigastric rising sensation/déjà vu leading to impaired awareness and automatisms) with or without secondary generalization on the basis of the hippocampal sclerosis, quite often after an early childhood event before the age of 5 years (eg, a febrile convulsion, meningitis, or head trauma).
Focal epilepsy often is further specified according to the localization of the presumed individual epileptogenic area (eg, temporal, frontal, parietal, occipital, insular) and can be either lesional or nonlesional based on CNS imaging.
Useful tests corroborating the clinical diagnosis and helping with further classification and determination of etiology:
1) Brain imaging studies: Computed tomography (CT) is the imaging modality used in the acute setting (eg, during the first seizure workup in the emergency room) helpful in excluding acute gross structural insults or traumatic sequelae. However, the imaging modality of choice to identify a distinct structural CNS lesion is magnetic resonance imaging (MRI). MRI should include a dedicated selection of sequences (“epilepsy protocol”; Table 12.3-2), acquired in optimal quality and, when possible, evaluated by neuroimaging specialists. Depending on the patient and the scanning protocol employed, ~30% of patients are “imaging negative,” which means that no structural abnormality causative for the clinical presentation can be identified. Furthermore, IGEs by definition do not show MRI lesions, whereas pharmacoresistant focal epilepsy shows relevant findings in up to 90% of patients with appropriate imaging studies.
2) EEG: Outpatient routine EEG is easily obtained and usually records 20 minutes of interictal EEG. Routine EEG often appears normal or displays only nonspecific abnormalities in patients with seizures, because specific diagnostic elements (eg, spike or sharp waves) are not necessarily frequent and therefore can be missed. Recording of a pathognomonic seizure event during a routine EEG is even less likely; however, recording of specific features, such as generalized or focal interictal epileptiform discharges, can be of great value. The sensitivity of EEG can be increased by a short interval between obtaining the EEG and the preceding seizure, repetitive investigation, applying trigger maneuvers (hyperventilation, sleep deprivation, photic stimulation), and including sleep stages. If a diagnosis remains uncertain, long-term EEG recording supplemented with video recording in a specialized inpatient unit (video-EEG unit) should be considered if available.
3) Cerebrospinal fluid (CSF) analysis: CSF testing is necessary when infectious or inflammatory CNS diseases are considered as possible etiologies. In recent years, new autoimmune mechanisms of epileptogenesis have been described and testing for autoantibodies in CSF samples can be helpful in selected cases of chronic seizure disorders. CSF analysis can also aid in the diagnosis of rare conditions such as glucose transporter type 1 deficiency by providing meaningful data (eg, serum/CSF glucose quotients). These tests usually require specialized consultations.
4) Laboratory testing: Laboratory testing can be helpful in assessing patients for metabolic disorders, which may have precipitated an ASS in the acute care setting (eg, hyponatremia, hypoglycemia, hyperglycemia, calcium level abnormalities), but it is of little use in most etiologic considerations of chronic epilepsy. However, laboratory investigations are important as an adjunct to monitoring response to pharmacotherapy, including serum levels of selected antiepileptic drugs or common therapy-induced adverse effects (eg, hyponatremia, hyperammonemia).
Diagnosing seizure complications: A useful part of physical examination in all patients is inspection of the tongue for tongue bites, which may be severe. Radiography and CT scanning are required to recognize seizure-related fractures and intracranial sequelae; note that fractures can be induced simply by the muscle force exerted during a seizure. Classic presentations are thoracic/thoracolumbar fractures and posterior shoulder luxation. Similarly, contractions can injure the structural integrity of muscle fibers and lead to significantly increased creatine kinase (CK)/myoglobin levels in the serum (rhabdomyolysis), which poses a risk to kidney function.
Differential diagnosis of seizures and epilepsy is extensive. Alternative diagnoses to be considered can be grouped into 3 categories: neurologic, nonneurologic medical, and psychiatric (Table 12.3-3). Specific etiologies include cardiovascular disease, movement disorders, metabolic disease, aberrations of the sleep-wake cycle, psychiatric disease, psychosomatic disease, and many others (eg, neurologic mimics, such as migraine and transient ischemic attack).
A common clinical presentation that must be distinguished from an epileptic seizure is syncope (see Syncope and Other Causes of Transient Loss of Consciousness). Major forms of syncope are related to CNS hypoperfusion (eg, due to cardiac dysfunction). It is not widely appreciated that in many instances transient global hypoperfusion of the CNS manifests as a loss of consciousness and motor signs (jerks, tonic signs)—so-called convulsive syncope. Another important condition that can mimic an epileptic seizure is the psychogenic nonepileptic seizure (PNES). PNESs are not produced by electrical CNS discharges but represent psychosomatic phenomena. Not voluntary and not simulated, they are behavioral attacks in the setting of a psychological dysfunction.
TreatmentTop
Acute treatment of a single seizure is rarely necessary because of the self-limiting character of most seizures, including TCSs. However, if considered necessary, standard benzodiazepine doses can be safely administered (eg, lorazepam 2 mg IV or orally). In patients with an ASS in the setting of an actual CNS insult, a standard antiepileptic drug is often chosen to suppress further seizures (eg, oral levetiracetam 500-1000 mg bid is often suitable; phenytoin is still in use but has its disadvantages); this could be considered as a transient management step and usually the consulting neurologist guides the length of treatment.
The first-line treatment of epilepsy is antiepileptic drugs (Table 12.3-4). Antiepileptic drugs, better referred to as antiseizure drugs (ASDs), encompass several pharmacologic classes of substances that act to suppress the aberrant electrical neuronal discharges. These agents have various mechanisms of action. Examples of common modern ASDs include levetiracetam, lamotrigine, carbamazepine/oxcarbazepine, lacosamide, topiramate, zonisamide, and perampanel. First-generation ASDs include phenytoin, phenobarbital, and valproic acid.
When choosing an ASD it is important to consider the type of epilepsy, adverse effect profile, and pharmacology of each agent, as well as the patient’s wishes regarding the appropriate monotherapy or combination therapy. Recently, lamotrigine and levetiracetam have been favored as the first-line choice in focal epilepsy, replacing the previously preferred carbamazepine. In IGEs, valproic acid is still considered a very effective ASD, although its use decreases because of the increasing awareness of potential adverse effects and due to medical and legal issues related to its teratogenicity. ASDs have varying modes of action and achieve their desired effect through selective ion channel blockade (eg, carbamazepine is a sodium channel blocker), targeting synaptic proteins (eg, levetiracetam binds to synaptic vesicle glycoprotein 2A), or action on postsynaptic receptors (eg, perampanel is an antagonist of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] receptors). It is important to note that these drugs do not alter or reverse the underlying epileptogenic process. They act by modulating transmission of electrical currents or by stabilizing membrane potentials and thus limiting neuronal excitability. About two-thirds of all ASD-treated patients with epilepsy are seizure free with medication alone. Pharmacoresistance can be declared when 2 regimens of an appropriately chosen ASD fail to control seizures; such cases warrant referral to an epilepsy center, which should include consideration of surgical intervention.
Common dose-dependent ASD adverse effects include drowsiness, dizziness, diplopia, nausea, vomiting, and cognitive slowing. Some ASDs can have rather substance-specific adverse effects, such as weight gain, hyponatremia, or psychiatric symptoms (Table 12.3-5). Certain drugs are teratogenic or potentially teratogenic and must be avoided in female patients of childbearing age. When available, the health-care provider should consult a specialist to select the appropriate drug. In patients with a first-time unprovoked seizure and those in whom the underlying cause has been adequately treated, long-term ASD administration is often unnecessary; conversely, most epilepsy disorders require life-long treatment with ASDs. The decision to taper off an ASD in a patient who achieved seizure freedom requires diligent counseling by an epilepsy specialist and is largely dependent on the specific epilepsy syndrome. Some forms of epilepsy are rather restricted to certain age groups (eg, childhood absence epilepsy, which can completely remit in adulthood), whereas others have high rates of relapse (eg, juvenile myoclonic epilepsy or lesional focal epilepsies).
Treatment of status epilepticus: see Status Epilepticus.
Surgical resection of a “CNS focus,” that is, the epileptogenic zone responsible for seizure generation, is a highly successful treatment option in focal epilepsy for carefully selected patients. Two-thirds of suitable patients who undergo epilepsy surgery achieve seizure freedom. The largest group of candidates for surgical intervention are those presenting with temporal lobe epilepsy, but many other epileptogenic targets can be treated safely. Each patient must be evaluated individually. Best results are achieved with complete removal of a well-demarcated epileptogenic lesion (eg, cavernous malformation, benign neoplasm, localized dysplasia). Comprehensive presurgical investigation and planning is critical to the safety and successful outcome of the operation, ensuring the greatest chance of seizure freedom and minimizing the risk of iatrogenic permanent neurologic deficits. Such expertise is offered by epilepsy centers only, and it remains of considerable concern that epilepsy surgery remains underused. Poor referral rates and delayed referral patterns mean that patients may have recurring seizures for as long as 20 years on average before they are assessed for epilepsy surgery.
Stimulation methods have been developed that may significantly reduce the frequency and/or intensity of seizures (eg, vagal nerve stimulation, deep brain stimulation, responsive neurostimulation) in patients with refractory epilepsy who are not good candidates for resective brain surgery. Such patients should be appropriately counseled that these methods rarely achieve seizure freedom. Possibilities include radiation (eg, stereotactic ablative radiosurgery, focal stereotactic low-dose radiation) and other less invasive methods (eg, laser ablation via interstitial thermocoagulation of electrically active foci or focused ultrasonography).
Psychosocial Treatment and Counseling
Caring for patients with epilepsy requires a multidisciplinary team of clinicians and allied health-care professionals to assist in coping with the psychosocial challenges that patients face as a result of their illness.
A wide range of psychiatric comorbidities are seen in patients with epilepsy (eg, depression, anxiety) that require careful attention and screening so that appropriate management and treatment can be implemented. Patients may also benefit from professional help in many other aspects of their life, including psychosocial support for completing their education, finding employment, and obtaining a driver’s license or living without a driver’s license. Others require support for activities of daily living or assisted living. Driving is usually restricted in most countries until a period of sustained seizure freedom is achieved and well documented; in many countries this period is defined as 1 year of seizure freedom, but specific regulations vary widely across countries and a review of the applicable legal principles is necessary. Counseling on pregnancy and parenthood is important.
Tables and FiguresTop
|
Disturbances of homeostasis |
– Glucose – Sodium – Calcium – Osmolality |
|
Drugs/drug withdrawal (selected agents) |
– Antidepressants (eg, amitriptyline, bupropion, clozapine) – Antibiotics (eg, penicillins, quinolones) – Immunotherapeutics (eg, cyclosporine, methotrexate) – Benzodiazepines/barbiturates – Alcohol, illicit drugs |
|
Acute illness |
– Renal failure – Liver failure – Hypertensive encephalopathy – Sepsis – Eclampsia – Porphyria |
|
Acute neurologic illness |
– Inflammatory (eg, meningitis, encephalitis, abscess) – Vascular – Acute trauma |
|
1. General requirements: High resolution, high contrast, complete brain coverage, optimal and thin slicing 2. Imaging can be achieved with a 2D protocol: – T1, T2, FLAIR: Both coronal and axial planes, ≥3 mm – T2*/SWI; contrast in selected patients – At least for TLE: Angulation along/perpendicular to the hippocampal axis 3. Most recent recommendations (if available): 3D sequences with isotropic voxels 1×1×1 in T1 and FLAIR: – No need for operator-dependent angulation – Reformatting in any plane possible |
|
FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging; SWI, susceptibility-weighted imaging; TLE, temporal lobe epilepsy. |
|
General category |
Seizure phenomena mimicked |
Leading symptom(s) |
|
Neurologic causes | ||
|
Movement disorders: – Dyskinesias – Episodic ataxia – Myoclonus – Startle reaction as a result of sudden surprise or alarm – Tic – Cataplexy |
– Motor seizures – Myoclonic seizures – Reflex seizures |
Motor signs |
|
Parasomnias: – NREM disorder – REM disorder – Narcolepsy |
– Nocturnal seizures – Negative motor seizure |
Sleep phenomena |
|
Other neurologic: – Migraine – TIA – Vertigo – Charles Bonnet syndrome |
– Posterior cortex seizure (parieto-occipital) |
– Somatosensory – Vertigo – Headache – Visual |
|
Nonneurologic medical causes | ||
|
Cardiovascular: – Rhythmogenic – Vasovagal – Orthostatic/POTS – Pulmonary embolism – Other (eg, carcinoid) |
Seizure with ictal asystole |
– Loss of consciousness – Syncope |
|
Metabolic: – Hypoglycemia – Intoxication – Tetany |
– Postictal state – Nonconvulsive SE |
Coma |
|
(Neuro-)psychiatric causes | ||
|
Dissociative and other: – Psychogenic nonepileptic seizure – Fugue – Psychosis – Stupor – PTSD – Panic attacks – Transient global amnesia – Organic disease affecting amnestic function – Dementia |
– Grand mal SE – Nonconvulsive SE – Amnesic seizure – Affective seizure – Focal SE |
– Behavioral symptoms – Altered mental state – Hallucinatory state – Amnestic state |
|
Physiologic phenomena | ||
|
A variety of normal phenomena could be mistaken for signs of disease, eg, myoclonic jerks that occur with transition to light sleep | ||
|
NREM, non–rapid eye movement; POTS, postural orthostatic tachycardia syndrome; PTSD, posttraumatic stress disorder; REM, rapid eye movement; SE, status epilepticus; TIA, transient ischemic attack. | ||
|
Drug |
Common daily dose (mg)a |
Common starting dose (mg)b |
First daily target dose (mg)c |
p450d |
Comments |
|
Carbamazepine |
600-1200 |
100-200 |
600-900 |
Inducer |
Self-induction at p450 |
|
Lamotrigine |
100-300 |
25 |
100 |
– |
Slow titration mandatory |
|
Levetiracetam |
1000-3000 |
250-500 |
1000-1500 |
– |
Excreted by kidneys |
|
Cenobamate |
100-400 |
12.5 |
100-150 |
Inhibition of CYP2C19 Induction of CYP3A4 |
New drug |
|
Lacosamide |
100-400 |
50-100 |
150-200 |
– |
Excreted by kidneys |
|
Oxcarbazepine |
600-1800 |
300 |
900-1200 |
Inducer |
Prodrug; converted to 10-OH-CBZ |
|
Perampanel |
6-12 |
2 |
4-6 |
– |
Very long half-life |
|
Topiramate |
50-200 |
25-50 |
100-150 |
Inducer |
Hepatic enzyme induction >200 mg |
|
Valproic acid |
900-2000 |
300-500 |
900-1200 |
Inhibitor |
Caution when adding to lamotrigine |
|
Zonisamide |
100-400 |
50 |
100-200 |
– |
Long half-life |
|
a The common daily dose refers to average levels. The upper value does not represent the maximum dose. b The common starting dose refers to usual starting doses per day. c The first daily target dose is a suggestion and refers to doses to be targeted by slow titration. d Agents with significant impact on drug metabolism by the liver. |
|||||
|
Common CNS adverse effects (dose dependent): Sedation, dizziness, blurred vision, diplopia, nausea, vomiting, ataxia, cognitive slowing | |
|
Possible substance-specific adverse effects (examples) | |
|
Carbamazepine |
Hyponatremia |
|
Lacosamide |
Prolongation of AV conduction |
|
Lamotrigine |
Insomnia, severe skin reactions, possible effects on ECG |
|
Levetiracetam |
Aggressive affect, irritability |
|
Oxcarbazepine |
Hyponatremia |
|
Phenytoin |
Gingival hyperplasia, hirsutism |
|
Pregabalin |
Weight gain |
|
Topiramate |
Weight loss, cognitive slowing, nephrolithiasis |
|
Valproic acid |
Weight gain, polycystic ovaries, hyperammonemia, thrombocytopenia, hair loss |
|
Zonisamide |
Nephrolithiasis, weight loss |
|
AV, atrioventricular; CNS, central nervous system; ECG, electrocardiography. | |
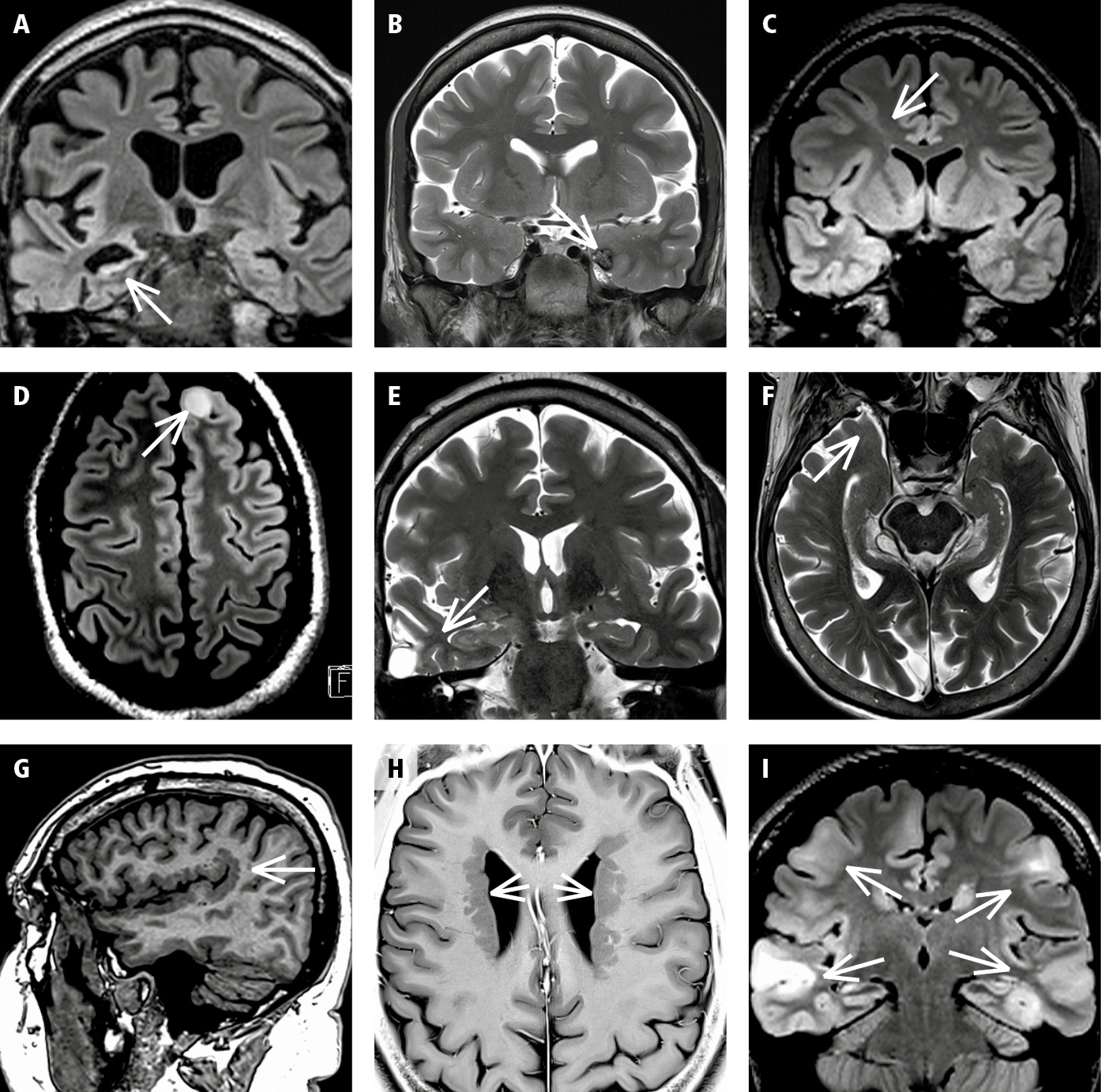
Figure 12.3-1. Selected classic structural lesions in epilepsy. A, hippocampal sclerosis; B, cavernoma; C, focal cortical dysplasia; D, neoplasia (benign tumor); E, traumatic scar; F, encephalocele; G, polymicrogyria; H, periventricular heterotopia; I, tuberous sclerosis.