Garmes HM, Boguszewski CL, Miranda PAC, et al. Management of hypopituitarism: a perspective from the Brazilian Society of Endocrinology and Metabolism. Arch Endocrinol Metab. 2021 Nov 1;65(2):212-230. doi: 10.20945/2359-3997000000335. Epub 2021 Feb 24. PMID: 33905631.
Higham CE, Johannsson G, Shalet SM. Hypopituitarism. Lancet. 2016 Nov 12;388(10058):2403-2415. doi: 10.1016/S0140-6736(16)30053-8. Epub 2016 Mar 31. Review. PubMed PMID: 27041067.
Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML; Endocrine Society. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011 Jun;96(6):1587-609. doi: 10.1210/jc.2011-0179. PubMed PMID: 21602453.
Definition, Etiology, PathogenesisTop
Hypopituitarism refers to a syndrome caused by deficiency of ≥1 pituitary hormone, which can result from pituitary or hypothalamic disease (deficiency of pituitary hormone–releasing hormones). Deficiency of all pituitary hormones is known as panhypopituitarism. The resulting changes in hormonal levels and in the choice of diagnostic tests are related to the presence of several short and long feedback loops among different parts of the hypothalamic-pituitary-peripheral gland system (Figure 1).
Causes: Table 1.
In general, the acquired loss of pituitary function follows the sequence of growth hormone (GH), luteinizing hormone (LH)/follicle-stimulating hormone (FSH), thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), and prolactin with tumors and radiation, but the sequence may vary with other etiologies.
Clinical Features and Natural HistoryTop
Clinical features of hypopituitarism vary depending on the cause, patient’s age, speed of onset, affected pituitary hormones, and magnitude of hormone deficiency, and they are often nonspecific at early stages (Table 2). Usually the deficiencies are permanent, but occasionally recovery may occur. Symptoms related to the underlying cause of hypopituitarism (sellar masses/tumors), such as bitemporal hemianopsia due to compression of the optic chiasm or diplopia due to invasion of the cavernous sinus, headache, and cerebrospinal fluid leakage may also be present. In the case of functioning pituitary tumors, symptoms resulting from increased hormone secretion may coexist. Additional specific situations are described below.
In patients with pituitary apoplexy (sudden hemorrhage into the pituitary gland), features may also include sudden-onset headache, nausea, vomiting, and confusion, which result from elevated intracranial pressure; visual disturbances resulting from compression of the optic chiasm and oculomotor nerves; paralysis of extraocular muscles caused by a hemorrhage to the cavernous sinus; and symptoms and signs of hypopituitarism. The most serious signs and symptoms of acute hypopituitarism are due to ACTH deficiency, which can result in life-threatening hypotension (refractory shock) and severe neurologic signs and symptoms in the case of a subarachnoid or intraventricular hemorrhage. Sometimes hemorrhagic apoplexy of a pituitary adenoma may result in spontaneous healing and subsequent resolution of symptoms of hormone hypersecretion.
Acute ACTH deficiency may occur when the pituitary gland is damaged in the course of pituitary apoplexy, stroke, head trauma, or neurosurgical procedures/surgery. It is a life-threatening condition (adrenal crisis: see Acute Adrenal Insufficiency). Chronic ACTH deficiency (see Central Adrenal Insufficiency) may become symptomatic and potentially lead to adrenal crisis after a sudden increase in the glucocorticoid requirements, especially in the setting of acute stress or infection.
Signs and symptoms of posttraumatic hypopituitarism usually develop gradually, becoming clinically overt within ~1 year after the event. In ~30% of patients severe head trauma may also cause damage to the hypothalamic supraoptic nucleus or the posterior pituitary lobe, resulting in antidiuretic hormone deficiency and the development of central diabetes insipidus (DI).
DiagnosisTop
Diagnosis entails a clinical examination looking for clinical manifestations of hormone deficiency (Table 2) and requires the documentation of subnormal secretion of pituitary hormones in basal and/or stimulated tests exploring stimulation and feedback loops in the endocrine system (Figure 1). Each pituitary hormone must be tested.
1. Basal laboratory studies:
1) Basal cortisol (early morning or 8:00 am) levels.
Pregnancy and medications such as combined hormonal contraceptives can cause falsely elevated serum cortisol levels.
2) TSH and free thyroxine (FT4) levels (FT4, not TSH, is the best marker of central hypothyroidism).
3) Men: LH and basal testosterone (morning, fasting; ideally 2 separate measurements are needed at separate times). Note that bioavailable testosterone testing is more accurate. Testosterone levels may be physiologically lower during periods of acute or subacute illness. Bioavailable testosterone should be measured in men with conditions that can affect sex hormone–binding globulin levels (eg, hypothyroidism and hyperthyroidism, obesity, diabetes mellitus, nephrotic syndrome, chronic liver disease, HIV infection, supraphysiologic glucocorticoid use/Cushing syndrome, acromegaly, GH deficiency, androgens, estrogens, anticonvulsant use).
Women: In patients with normal menses, no tests are needed; if menses are abnormal, measure the levels of FSH, LH, and estradiol. Combined hormonal contraception use in women may lead to suppressed FSH, LH, and estradiol levels.
4) Insulin-like growth factor 1 (IGF-1; also called somatomedin C; its production is stimulated by GH). GH levels are not helpful for diagnosing GH deficiency due to the pulsatile secretion of GH.
5) Prolactin.
6) Serum sodium, serum and urine osmolality if signs and symptoms of DI are present: these may be normal if the patient has free access to water and more definitive testing may be required, such as a water deprivation test.
2. Magnetic resonance imaging (MRI) of the parasellar region should be performed in every case of hypopituitarism to look for a sellar lesion.
3. Visual fields should be assessed in patients with suspected pathologic lesions in the area of the optic chiasm or visual field deficits on examination.
1. Hypothalamic-pituitary-adrenal axis: see Primary Adrenal Insufficiency.
2. Thyroid function: TSH and FT4. Low FT4 levels with normal, low, or slightly elevated TSH levels (<10 IU/L) are consistent with central hypothyroidism.
3. Gonadotropins:
1) Men: Total fasting testosterone and LH levels (measured in the morning). A low testosterone level with a normal or low LH level is consistent with hypogonadotropic hypogonadism. Note that bioavailable testosterone is more accurate in certain clinical scenarios, as described above.
2) Women: In patients with normal menses, no tests are needed. In patients with abnormal menses, measure the levels of FSH, LH, and estradiol. Low estradiol concentrations with normal or low FSH and LH levels are consistent with hypogonadotropic hypogonadism.
4. GH:
1) IGF-1: A concentration lower than the age-specific lower limit of a normal range in a patient with pituitary disease with multiple pituitary deficiencies confirms GH deficiency (IGF-1 production is stimulated by GH).
2) Provocative dynamic tests: Insulin-induced hypoglycemia, glucagon stimulation test, or arginine with growth hormone–releasing hormone (GHRH) stimulation test. Subnormal increases of GH here are consistent with GH deficiency.
5. Prolactin: Patients with low prolactin concentrations usually have no manifestations. An exception are women in the postpartum period, who may not be able to breastfeed (see Hyperprolactinemia).
6. Antidiuretic hormone deficiency: If history is consistent with DI, serum sodium levels are normal, and serum and urine osmolality is equivocal, consider a water deprivation test [see Diabetes Insipidus]).
Differential diagnosis depends on the signs and symptoms and will vary, depending on the pituitary deficiencies, but may include primary adrenal insufficiency, primary hypothyroidism, or primary hypogonadism. Nevertheless, measurements of biochemical hormone levels, basal and/or stimulated, will confirm a definite diagnosis.
TreatmentTop
Management of hormone deficiencies in central hypopituitarism involves the administration of appropriate target hormones.
1. Central adrenocortical insufficiency: Glucocorticoids. Typically start hydrocortisone 15 to 25 mg/d in 2 to 3 divided doses. An alternative is prednisone 5 to 7.5 mg daily (see Central Adrenal Insufficiency).
2. Central hypothyroidism: Levothyroxine in individually adjusted doses (first correct the adrenocortical insufficiency to avoid adrenal crises). Increase the dose gradually, for instance, starting with 25 microg daily in older patients or in those with cardiac disease and titrating the dose up based not on TSH levels but on the patient’s clinical condition and FT4 levels to aim for a mid-high normal FT4. In younger patients without cardiac conditions, a replacement dose of levothyroxine of 1.6 microg/kg/d can be initiated and then adjusted as above. FT4 levels should be checked 6 to 8 weeks after initiation and dose change. Pregnant patients generally do not require an increase in levothyroxine due to preservation of the thyroid response to the thyroid-stimulating effect of human chorionic gonadotropin, beta subunit (beta-hCG; unlike in primary hypothyroidism, where increased doses of levothyroxine are usually required).
1) Men, active fertility is not an issue: Testosterone.
a) IM/subcutaneous testosterone enanthate or cypionate 100 mg weekly or 200 mg every 2 weeks, or undecanoate 750 mg; repeat the 750-mg dose after 4 weeks and then every 10 weeks.
b) Topical transdermal testosterone (50-100 mg of 1% gel; 20.25-81 mg of 1.62% gel, or 40-70 mg of 2% gel daily to skin or a 2.5-5 mg patch daily).
c) Oral (testosterone undecanoate 40-120 mg/d, bid to tid, in divided doses with meals – this formulation is less effective and not preferred).
d) Target serum testosterone levels to upper-middle part of the normal range; also monitor the prostate-specific antigen (PSA) and perform digital rectal examination (DRE) as clinically indicated along with benign prostatic hyperplasia (BPH) and polycythemia testing.
2) Men, fertility is desired: Gonadotropins (LH and FSH), gonadotropin-releasing hormone therapy, hCG, clomiphene citrate, or aromatase inhibitors in certain cases.
Note that testosterone replacement should not be given to patients with prostate/breast cancer, uncontrolled heart failure, severe untreated BPH, moderate to severe untreated sleep apnea, recent myocardial infarction (MI)/stroke, elevated PSA, thrombophilia, palpable prostate nodule or induration, and polycythemia/erythrocytosis (hematocrit ≥54%).Evidence 1Strong recommendation (benefits clearly outweigh downsides; right action for all or almost all patients). Low Quality of Evidence (low confidence that we know true effects of the intervention). Quality of Evidence lowered due to risk of bias. Breast and prostate cancers are hormone-dependent cancers, and low-quality evidence (risk of bias) suggests that testosterone may cause tumor growth. The recommendation against testosterone replacement places a high value in avoiding a potential catastrophic adverse effect. Fowler JE Jr, Whitmore WF Jr. The response of metastatic adenocarcinoma of the prostate to exogenous testosterone. J Urol. 1981 Sep;126(3):372-5. PubMed PMID: 7277602.
3) Women: In women with an intact uterus, use low-dose estrogens (oral estradiol 0.5-2 mg daily, oral conjugated equine estrogen 0.3-1.25 mg daily, or transdermal estradiol patch [changed once or twice weekly to give a 25-100 microg daily equivalent] or gel 1.5 mg daily) combined with cyclic or continuous progestins to prevent endometrial hyperplasia (oral micronized progesterone 200 mg for 7-12 days of the month for cyclic dosing or 100 mg daily for continuous dosing; or oral medroxyprogesterone acetate 5 mg for 7-12 days of the month for cyclic dosing or 2.5 mg daily for continuous dosing). Other alternative progesterones include norethindrone (0.35 mg), gestodene (0.75 mg), or levonorgestrel (0.075 mg). Combined estrogen-progestin contraceptives may be considered and more acceptable for younger women. Regardless, estrogen-progestin therapy should be used at the lowest effective dose to control hypoestrogenic symptoms, protect bones, and prevent adverse cardiovascular effects. The dose should be reduced with advancing age.
In patients without a uterus, use estrogen replacement therapy alone without progestins.
Absolute contraindications to estrogen therapy include the presence of hormone-sensitive cancers (breast cancer, endometrial cancer), use of estrogen therapy alone (without progesterone) in patients with an intact uterus, previous venous thromboembolism (VTE) or stroke/transient ischemic attack (TIA), active liver disease, migraine with aura.
If fertility restoration is a goal, potential treatments may include clomiphene citrate, aromatase inhibitors, hCG, human menopausal gonadotropin, or purified or recombinant FSH.
4. GH deficiency in adults: Recombinant human GH. GH replacement therapy is also suggested in adults with severe GH deficiency and no contraindications.
Contraindications include active malignancy, proliferative or severe nonproliferative diabetic retinopathy, and critical illness. Patients need to be monitored for fluid retention, worsening glucose intolerance, and intracranial hypertension (especially in the first 8 weeks of treatment).Evidence 2Weak recommendation (benefits likely outweigh downsides, but the balance is close or uncertain; an alternative course of action may be better for some patients). Moderate Quality of Evidence (moderate confidence that we know true effects of the intervention). Quality of Evidence lowered due to inconsistency among different outcomes measured, which suggests that therapy in adults with confirmed GH deficiency reduces their weight and body fat and increases lean body mass but can worsen peripheral edema and joint stiffness. This recommendation is also weak rather than strong due to the high cost and burden of treatment. Hazem A, Elamin MB, Bancos I, et al. Body composition and quality of life in adults treated with GH therapy: a systematic review and meta-analysis. Eur J Endocrinol. 2012 Jan;166(1):13-20. doi: 10.1530/EJE-11-0558. Review. PubMed PMID: 21865409.
5. Central DI: Desmopressin/DDAVP, available as:
1) Oral formulation: Start with 0.05 to 0.2 microg at bedtime; the maintenance dose is typically 0.1 to 0.8 microg daily in divided doses.
2) Sublingual formulation: Start with 60 microg tid; the maintenance dose is typically 120 to 720 microg daily in divided doses.
3) Intranasal formulation: Start with 5 to 10 microg daily at bedtime; the maintenance dose is typically 5 to 20 microg daily in divided doses.
4) IV or subcutaneous formulations can be used for inpatients (see Diabetes Insipidus).
Interactions between different types of hormone replacement therapy: Table 3.
Treatment of Underlying Conditions
1. Tumors in the pituitary:
1) Surgical resection (transsphenoidal) is the first-line treatment for most pituitary adenomas (except for prolactinoma [see below]), many other sellar lesions/tumors, tumors derived from Rathke pouch, and other parasellar tumors (except for germinomas).
2) Pharmacotherapy: Dopamine agonists for prolactinoma (cabergoline, quinagolide, and bromocriptine). In TSH-secreting pituitary adenomas, long-acting somatostatin analogues are used to restore euthyroidism prior to the definitive treatment with transsphenoidal surgery. In the case of GH-secreting tumors, the routine use of pharmacotherapy (somatostatin analogues) as pretreatment before the neurosurgical resection is usually not used except in patients with significant pharyngeal thickness, severe sleep apnea, very invasive tumors, high-output heart failure, or delayed surgery.
3) Radiotherapy is indicated for germinoma as primary therapy and should be considered for other unresectable tumors, either primary or recurring/aggressive/persistent after neurosurgery or, if appropriate, refractory or intolerant to medical treatment. Modern radiotherapy (stereotactic) approaches are associated with a lower risk of complications.
4) Chemotherapy is used in the case of metastatic tumors of the pituitary or as an adjuvant treatment in patients with central nervous system tumors sensitive to chemotherapy.
2. Hemorrhagic pituitary apoplexy:
1) IV glucocorticoids: Hydrocortisone 100 mg qid to tid or dexamethasone ~4 mg bid at the early stages of pituitary apoplexy to correct the possible ACTH deficiency and reduce vasogenic edema.
2) Surgical decompression: The decision to proceed to surgery usually should be made within 1 week from hemorrhagic apoplexy in patients whose neurologic status does not improve despite the administration of glucocorticoids (and, in the case of prolactinoma, possibly dopamine agonist therapy).
PrognosisTop
Adequate hormone replacement therapy allows maintaining a good overall clinical status. However, the mortality rates are higher compared with the general population, regardless of the cause of pituitary insufficiency, and quality of life is impaired. Specific attention is required towards long-term cardiovascular disease risks and long-term risk of metabolic comorbidities, which are increased, and to glucocorticoid replacement at the time of acute illness (‘stress dose’). The prognosis is less favorable in patients with the development of central DI and in those with malignant central nervous system tumors causing pituitary insufficiency, and depends on the type and stage of the tumor. With certain etiologies of hypopituitarism, some hormone deficiencies may be temporary and may improve over time, so retesting should be considered periodically.
Tables and FiguresTop
| Cause | Details |
|
Tumors |
Pituitary adenomas (functioning and nonfunctioning), craniopharyngiomas, posterior pituitary tumors (ganglioneuroma, astrocytoma), hypothalamic tumors (astrocytoma, germinoma), tumors of the skull base/optic chiasm (meningioma, glioma), metastases (most frequently breast and lung cancer) |
|
Cystic lesions |
Rathke cleft, arachnoid, epidermoid cysts |
|
Cranial trauma and iatrogenic |
Surgery (most frequently transsphenoidal sellar lesion), severe head trauma/brain injury, pituitary/sellar irradiation |
|
Vascular |
Postpartum pituitary infarction (due to postpartum hemorrhage [Sheehan syndrome]), pituitary apoplexy (sudden hemorrhage into the pituitary gland), internal carotid artery aneurysm (compression), stroke (ischemic or subarachnoid hemorrhage) |
|
Inflammatory and infiltrative lesions |
Sarcoidosis, hemochromatosis, Langerhans cell histiocytosis, granulomatosis with polyangiitis (formerly Wegener’s disease), hypophysitis related to immune checkpoint inhibitors, autoimmune, granulomatous, xanthomatous, or IgG-4 related disease |
|
Drug/medication related |
Supraphysiologic corticosteroids, high-dose opiates |
|
Infections |
Tuberculosis, syphilis, mycoses, pituitary abscess, encephalitis, meningitis |
|
Congenital (rare) |
Familial hypopituitarism with multiple hormone deficiencies (PROP1, HESX1, PIT1) or developmental abnormalities (pituitary hypoplasia or aplasia) |
|
Isolated hormone deficiencies |
– Defective gonadotropin-releasing hormone synthesis (causing hypogonadotropic hypogonadism) associated with hyposmia, functional disorders, and certain medications – Isolated deficiency of ACTH, TSH, or prolactin (very rare) – Transient ACTH deficiency possible in the setting of prolonged higher-than-physiological glucocorticoid replacement |
|
a The most common causes of hypopituitarism are in boldface. |
|
|
ACTH, adrenocorticotropic hormone; TSH, thyroid-stimulating hormone. |
|
|
Deficiency |
Signs and symptoms |
|
GH |
Growth retardation (in children), decreased muscle mass, increased body fat (primarily visceral fat), decreased bone mineral density, hypoglycemia, dyslipidemia, poor QOL and wellbeing (in adult-onset deficiency, signs and symptoms are nonspecific) |
|
ACTH |
Orthostatic hypotension, presyncope/syncope, nausea and vomiting, anorexia, weight loss, decreased skin pigmentation, tendency to hypoglycemia (particularly in patients with coexisting GH deficiency) |
|
TSH |
Central hypothyroidism (signs and symptoms are similar but less pronounced than in primary hypothyroidism, goiter is absent) |
|
LH and FSH |
Amenorrhea, male sexual dysfunction, infertility, decreased libido, regression or lack of secondary sexual characteristics (pubic hair), decreased bone mineral density |
|
PRL |
No lactation after childbirth |
|
ACTH, adrenocorticotropic hormone; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; PRL, prolactin; QOL, quality of life; TSH, thyroid-stimulating hormone. | |
|
Glucocorticoids |
Thyroxine |
GH |
|
|
Thyroxine |
Assess HPA axis before starting thyroxine, as starting thyroxine in patients with adrenal insufficiency can precipitate an adrenal crisis |
— |
GH therapy can lower FT4 levels, so increase of the dose of levothyroxine may be needed |
|
Estrogen |
Oral estrogens increase cortisol-binding globulin levels, which can cause elevated serum cortisol (not seen with transdermal estrogen) |
Estrogen increases thyroid binding globulin levels, so initiation or increase of the dose of levothyroxine may be needed |
Oral estrogen lowers serum IGF-1 levels, so higher doses of GH therapy may be required (this is not seen with transdermal estrogen) |
|
GH |
GH decreases conversion of cortisone to cortisol, so higher doses of glucocorticoids may be required |
— |
— |
|
ADH |
AI can mask DI and treatment of AI can unmask DI |
— |
— |
|
—, not applicable. |
|||
|
ADH, antidiuretic hormone; AI, adrenal insufficiency; DI, diabetes insipidus; FT4, free thyroxine; GH, growth hormone; IGF-1, insulin-like growth factor 1. |
|||
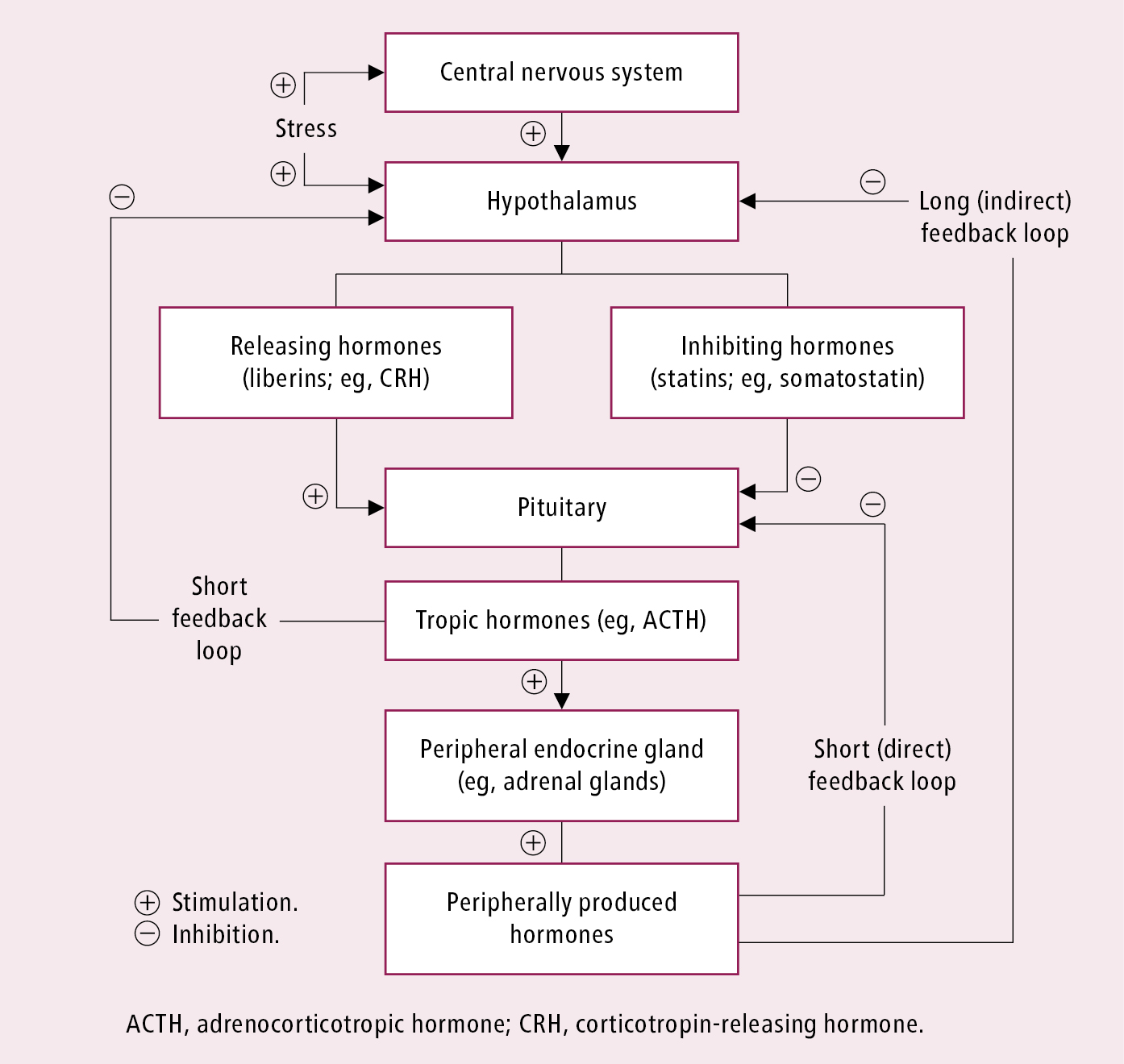
Figure 6.3-1. Stimulation and feedback loops in the endocrine system.
 English
English
 Español
Español
 українська
українська