Definición y etiopatogeniaArriba
El mieloma múltiple (MM) es una enfermedad neoplásica de curso gradual caracterizada por la proliferación descontrolada, multifocal y el acúmulo de plasmocitos monoclonales en la médula ósea que producen una inmunoglobulina monoclonal o cadenas ligeras monoclonales de inmunoglobulinas (denominada proteína M) y provocan daños en los órganos. La etiología de la enfermedad es desconocida. La edad promedio de aparición de la enfermedad es de 70 años.
Cuadro clínico e historia naturalArriba
1. Las manifestaciones clínicas se deben a la expansión de los plasmocitos neoplásicos, y a las proteínas monoclonales anormales y citoquinas secretadas por estos:
1) Signos y síntomas generales: debilidad y pérdida de peso.
2) Dolor óseo (es el síntoma más frecuente): localizado a nivel lumbar, de la pelvis, o de las costillas, raramente en el cráneo o en los huesos largos. Está causado por lesiones osteolíticas y fracturas óseas patológicas (p. ej. fracturas vertebrales por compresión).
3) Manifestaciones neurológicas: se producen a consecuencia de la compresión o lesión de la médula espinal, de las raíces de los nervios espinales o de los nervios craneales por fracturas patológicas (p. ej. de vértebras) o directamente por el tumor. Con mayor frecuencia radiculopatías, a veces paresias, parálisis de extremidades, incontinencia urinaria o fecal. La polineuropatía periférica sensorial o sensitivomotora, simétrica y distal, es rara en el momento de diagnóstico, más frecuente en enfermos con coexistencia de amiloidosis de cadenas ligeras de inmunoglobulinas y con el síndrome POEMS, así como en pacientes tratados con fármacos neurotóxicos (talidomida, bortezomib).
4) Signos y síntomas de anemia (~70 %) →Anemias.
5) Signos y síntomas de hipercalcemia y sus consecuencias →Hipercalcemia.
6) Infecciones recurrentes bacterianas del sistema respiratorio y de las vías urinarias, e infecciones virales (gripe, herpes zóster).
7) Signos y síntomas de insuficiencia renal: se describen en ~30 % de los pacientes en el momento de diagnóstico de MM. Con mayor frecuencia es la denominada nefropatía por cilindros cuya presentación clínica es la lesión renal aguda (nefritis tubulointersticial por cilindros intratubulares de cadenas ligeras en la orina).
8) Manifestaciones del síndrome de hiperviscosidad (en <10 % de los enfermos): más frecuentemente diátesis hemorrágica (epistaxis, gingivorragia, púrpura), empeoramiento de la agudeza visual, síntomas de compromiso del SNC (cefalea, sordera súbita, vértigo, ataxia, nistagmo, alteraciones de la conciencia), agudización de la insuficiencia cardíaca.
9) Más raramente: plasmocitomas extramedulares, signos y síntomas de amiloidosis AL concomitante, hepatomegalia, adenopatías periféricas, esplenomegalia, síndrome de Fanconi.
2. Historia natural: en ~10-15 % de los enfermos el curso es benigno (mieloma asintomático).
DiagnósticoArriba
Exploraciones complementarias
1. Hemograma de sangre periférica: en la mayoría de los enfermos se describe anemia normocítica, normocrómica, con menor frecuencia macrocitosis, en un 50 % de los enfermos eritrocitos en pila de monedas, y más raramente leucopenia y trombocitopenia.
2. Estudio de proteínas en suero y en orina: hiperproteinemia, hipergammaglobulinemia monoclonal, disminución de inmunoglobulinas normales, presencia de proteína M en la electroforesis e inmunofijación en el suero y orina (en el mieloma de cadenas ligeras [~20 %] en general se observa una panhipogammaglobulinemia en lugar de la imagen típica en la electroforesis de proteínas séricas), elevación de cadenas ligeras libres monoclonales (sFLC: κ o λ) en sangre y/u orina (proteína de Bence Jones en orina), con una relación anormal de concentraciones κ/λ.
3. Aspirado y biopsia de médula ósea: aumento del porcentaje de plasmocitos monoclonales.
4. Estudio citogenético para determinar el grupo de riesgo (→tabla 16.15-1).
5. Otras pruebas de laboratorio: VHS elevada (a menudo superior a 100), hipercalcemia, elevación de ácido úrico, creatinina, β2-microglobulina, proteína C-reactiva en el suero, y de LDH en el suero, raramente crioglobulinemia.
6. Pruebas de imagen óseas (radiografía, TC y/o RMN o PET-TC): focos osteolíticos (principalmente en huesos planos y largos), osteopenia y osteoporosis, fracturas patológicas. Las radiografías deberían incluir el cráneo, los húmeros, los fémures, la pelvis, columna vertebral y las zonas dolorosas. En función de la disponibilidad, se debe realizar un examen de mayor sensibilidad: TC de baja radiación de cuerpo entero, eventualmente RMN o PET-TC. La RMN (eventualmente la TC) es la técnica de elección en caso de sospechar fracturas compresivas o compresión de la médula espinal, o cuando la radiografía no muestra cambios patológicos en áreas sintomáticas.
Criterios diagnósticos
1. MM: plasmocitos clonales en médula >10 % o plasmocitoma óseo o extramedular confirmado por biopsia junto con ≥1 de los siguientes criterios:
1) Criterios de afectación orgánica relacionados con el mieloma (CRAB):
a) hipercalcemia (>0,25 mmol/l por encima del LSN o >2,75 mmol/l)
b) insuficiencia renal (aclaramiento de creatinina <40 ml/min o creatininemia >177 µmol/l [2 mg/dl])
c) anemia (Hb 2 g/dl por debajo del LIN o <10 g/dl)
d) lesiones óseas (≥1 foco osteolítico en radiografía, TC o PET-TC).
2) ≥1 biomarcador neoplásico (SLiM):
a) plasmocitos en médula >60 %
b) relación de cadenas ligeras libres en el suero (κ/λ o λ/κ) >100 con una concentración de cadenas monoclonales >100 mg/l
c) >1 lesión focal de tamaño ≥5 mm en la RMN.
2. Mieloma asintomático (latente): proteína M en el suero ≥30 g/l o en orina >500 mg/24 h y/o plasmocitos clonales en médula 10-60 %, sin que se cumplan los criterios CRAB y SLiM, sin amiloidosis AL.
3. Mieloma no secretor: no se detecta proteína monoclonal en inmunofijación en suero y orina, aunque en 2/3 de los casos hay un aumento de la concentración de sFLC monoclonales y/o relación alterada de sFLC κ/λ.
4. Leucemia de células plasmáticas: recuento de plasmocitos neoplásicos en la sangre >2000/µl o >5 % de los leucocitos circulantes. Forma agresiva con mal pronóstico y corta supervivencia.
5. Plasmocitoma solitario: tumor único en hueso o extraóseo (en la mayoría de los casos en las vías respiratorias superiores), con resultado normal del examen de huesos (incluso en la RMN o en la TC de columna vertebral y de pelvis), sin manifestaciones de CRAB.
Diagnóstico diferencial
1) Otras gammapatías monoclonales (→Gammapatías monoclonales: información general, →tabla 16.15-2).
2) Plasmocitosis reactiva policlonal (reacción plasmocitaria): en el curso de infecciones, p. ej. rubéola, mononucleosis infecciosa, infecciones crónicas, enfermedades hepáticas (el porcentaje de plasmocitos en la médula ósea generalmente es <10 %, no hay proteína M).
3) Hipergammaglobulinemia policlonal.
4) Neoplasias que producen metástasis óseas: p. ej. cáncer renal y de mama, carcinoma pulmonar no microcítico, cáncer de próstata.
5) Insuficiencia renal de otra etiología, p. ej. en un enfermo con GMSI.
TratamientoArriba
Tratamiento antineoplásico
1. Enfermos <70 años o ≥70 años sin enfermedades concomitantes: el tratamiento de inducción se inicia con 3-4 ciclos de un esquema con tres fármacos que contenga bortezomib (eventualmente otro inhibidor de proteasoma), dexametasona y un 3.er fármaco, idealmente inmunomodulador (con mayor frecuencia VTD [talidomida], VCD [ciclofosfamida], VRD [lenalidomida]). La asociación de daratumumab (anti-CD38) a VTD mejora la eficacia del tratamiento y prolonga la supervivencia libre de progresión. A continuación, tras la movilización de células hematopoyéticas (G-CSF con o sin quimioterapia) se realiza quimioterapia con dosis altas (melfalán a dosis de mieloablación), consolidada con trasplante de células hematopoyéticas de sangre periférica autólogas (auto-PBSCT). Posteriormente debe evaluarse la posibilidad de administrar aún 2-3 ciclos del protocolo administrado antes del auto-PBSCT, o realizar un nuevo auto-PBSCT 3-6 meses después del primero.
2. Enfermos no aptos de auto-PBSCT (→tabla 16.15-2): quimioterapia, normalmente VRD, VMP (bortezomib + melfalán + prednisona), RD (lenalidomida + dexametasona), eventualmente VCD, VD (bortezomib + dexametasona).
3. Enfermos después del auto-PBSCT o después del tratamiento de inducción que no ha terminado en auto-PBSCT: tratamiento de mantenimiento de la remisión (lenalidomida, bortezomib, eventualmente talidomida).
4. Resistencia o recidiva de la enfermedad (→tabla 16.15-3): esquemas de 2 o 3 fármacos (con dexametasona), compuestos tanto por fármacos de primera línea (lenalidomida, bortezomib) y sus análogos más recientes (pomalidomida, carfilzomib e ixazomib) como por fármacos citostáticos tradicionales (bendamustina) y fármacos de otros mecanismos de acción (daratumumab, isatuximab, elotuzumab, belantamab mafodotin, panobinostat, selinexor). La elección de la terapia depende del tratamiento anterior, la duración de la respuesta, la edad, el estado general del enfermo, las enfermedades concomitantes, la disponibilidad de los fármacos y la dinámica de la enfermedad. En enfermos seleccionados: auto- o alo-PBSCT.
5. Mieloma asintomático (latente): vigilancia cada 3-6 meses.
6. Plasmocitoma solitario: cirugía o radiación. Seguimiento por riesgo de una eventual progresión hacia MM.
Tratamiento de soporte
1. Tratamiento de la enfermedad renal:
1) plasmaféresis o hemodiálisis high cut-off a considerar en la nefropatía por cilindros
2) hidratación adecuada del enfermo (≥3 l/d [≥2 l/m2/d]) al iniciar el tratamiento
3) inicio inmediato del tratamiento antineoplásico con inclusión en el primer ciclo de bortezomib y dexametasona a dosis altas (40 mg/d durante 4 días)
4) evitar fármacos nefrotóxicos (p. ej. AINE, aminoglucósidos, furosemida) y contrastes radiológicos
5) tratamiento de la hipercalcemia →Hipercalcemia e hiperuricemia →Síndrome de lisis tumoral y →Gota
6) adecuación de la dosis de algunos fármacos (lenalidomida, melfalán, ácido zoledrónico, heparina) al aclaramiento de creatinina
7) tratamiento de la LRA →Lesión renal aguda y de la ERC →Enfermedad renal crónica (ERC).
2. Inhibición de la osteólisis: utilizar los inhibidores de osteoclastos (bisfosfonatos o denosumab) en todos los enfermos con MM sintomático (también sin osteólisis). Bisfosfonatos:
1) ácido zoledrónico iv. 4 mg (3-3,5 mg, cuando el aclaramiento de creatinina 30-60 ml/min) en 100 ml de NaCl al 0,9 % en infusión de ≥15 min cada 3-4 semanas
2) pamidronato iv. cada 3-4 semanas 30 mg en 250 ml de NaCl al 0,9 % en infusión de ≥45 min o 90 mg en 500 ml de NaCl al 0,9 % en infusión de ≥2 h.
El fármaco de elección es el ácido zoledrónico. Los bisfosfonatos están contraindicados en enfermos con TFGe <30 ml/min/1,73 m2, excepto los en tratamiento crónico con hemodiálisis sin posibilidad de recuperar la función renal. El ácido zoledrónico se utiliza durante ≥12 meses hasta obtener al menos una respuesta parcial muy buena (RPMB; →tabla 16.15-4). Se utiliza pamidronato mientras el mieloma esté activo. Al tomar la decisión de suspender el tratamiento con bisfosfonatos, tener en cuenta la evaluación individual de riesgo de fracturas. En caso de progresión bioquímica o clínica de MM en pacientes que ya no reciben bisfosfonatos, se debe reinstaurar este tratamiento.
El denosumab VSc a dosis de 120 mg 1 × mes es una alternativa a los bisfosfonatos, sobre todo en enfermos con insuficiencia renal. Se utiliza durante ≥2 años y hasta conseguir ≥RPMB. En caso de suspender el tratamiento con denosumab, para evitar el efecto rebote se debe administrar después de 6 meses 1 dosis de bisfosfonato, o continuar la terapia con denosumab 1 × cada 6 meses.
Debido al riesgo de necrosis mandibular, antes de iniciar el tratamiento con inhibidor de osteoclastos es necesario realizar saneamiento de la cavidad oral. Durante esta terapia se recomienda suplementar VO calcio (tras normalizar una eventual hipercalcemia) y vitamina D, vigilar la función renal, los niveles de calcio y de otros electrólitos, cuidar la higiene bucal y evitar intervenciones odontológicas mayores (si son necesarias, se debe pausar el tratamiento).
3. Tratamiento de hipercalcemia y crisis hipercalcémica →Hipercalcemia.
4. Tratamiento del síndrome de hiperviscosidad: plasmaféresis con sustitución de albúmina o de plasma.
5. Tratamiento de la anemia →Anemia en enfermedades crónicas y/o →Anemia en la enfermedad renal crónica.
6. Tratamiento del dolor óseo →Dolor en el enfermo oncológico.
7. Prevención de infecciones:
1) vacunas contra gripe, neumococo, H. influenzae y COVID-19
2) considerar la administración de cotrimoxazol 960 mg 1 × d durante 3 d/semana o ciprofloxacina 500 mg 1 × d, o levofloxacina 500 mg 1 × d durante los primeros 2-4 meses del tratamiento, y en enfermos con hipogammaglobulinemia e infecciones bacterianas recurrentes
3) en enfermos con niveles disminuidos de inmunoglobulinas normales con infecciones recurrentes o graves a pesar de la profilaxis antimicrobiana y vacunación se debe considerar IGIV o IGSC
4) aciclovir o valaciclovir en enfermos tratados con inhibidor de proteasoma (bortezomib, carfilzomib, ixazomib) o con anticuerpo anti-CD38 (p. ej. daratumumab), o sometidos a auto-PBSCT, y a considerar en enfermos tratados con inmunomoduladores (p. ej. lenalidomida) con glucocorticoides a dosis altas y con antecedentes de herpes zóster
5) G-CSF en casos seleccionados.
8. Prevención antitrombótica: en enfermos tratados con esquemas basados en un fármaco inmunomodulador (talidomida, lenalidomida) debe administrarse AAS a dosis de 75-100 mg/d, HBPM (a dosis profiláctica →tabla 2.33-4), o warfarina (en enfermos con >1 factor de riesgo adicional de ETV).
9. Tratamiento de la polineuropatía secundaria a medicamentos: adecuación de la dosis, del modo de administración o discontinuación del fármaco neurotóxico (talidomida, bortezomib). Tratamiento del dolor neuropático →fig. 24.1-2.
PronósticoArriba
El tratamiento permite conseguir la remisión (→tabla 16.15-4) y prolongar la supervivencia. En la mayoría de los casos la enfermedad progresa o recidiva después de diversas líneas de tratamiento. Los resultados del tratamiento tras cada recidiva son peores. La causa de muerte más frecuente son las infecciones. La mediana de supervivencia en enfermos con MM sintomático y progresivo es de 5-7 años (→tabla 16.15-1).
TABLAS Y FIGURAS
Sistema de estadificación internacional (ISS) e ISS modificado (R-ISS) para el mieloma múltiple
|
ISS
|
|
Estadio
|
Criterios
|
Mediana de tiempo
de supervivencia
(meses)
|
|
β2-microglobulina
en suero (mg/l)
|
Albúmina
en suero (g/l)
|
|
1
|
<3,5
|
≥35
|
62
|
|
2
|
<3,5
|
<35
|
45
|
|
3,5-5,5
|
Independientemente
|
|
3
|
>5,5
|
Independientemente
|
29
|
|
R-ISS
|
|
Estadio
|
Criterios
|
ST a los 5 años
|
SLDP a los 5 años
|
|
1
|
ISS1, sin cambios citogenéticos de alto riesgoa y LDH en el suero normal
|
82 %
|
55 %
|
|
2
|
No se cumple el criterio R-ISS 1 o 3
|
62 %
|
36 %
|
|
3
|
ISS 3 y cambios citogenéticos de alto riesgoa o LDH en el suero >LSN
|
40 %
|
24 %
|
|
a En la hibridación fluorescente in situ (FISH) en interfase: del(17p) o t(4;14) o t(14;16).
LDH — lactato deshidrogenasa, SLDP — supervivencia libre de progresión, ST — supervivencia total
A partir de: J. Clin. Oncol., 2005; 23 (15): 3412 y J. Clin. Oncol., 2015; 32: 2173
|
Diagnóstico diferencial de discrasias plasmocitarias no-IgM
|
|
GMSI no-IgM
|
GMSR
|
EDCL
|
AL
|
POEMSa
|
Plasmocitoma solitario / plasmocitoma extraóseo
|
MMA
|
MM
|
Mieloma no secretor
|
Leucemia de células plasmáticas
|
|
Proteína M en suero o en orinab
|
<30 g/l en suero y <500 mg/24 h en orina
|
Habitualmente concentración baja (como en GMSI)
|
Ausente o concentración baja (como en GMSI)
|
≥30g/l en suero o >500 mg/24 h en orina y/o plasmocitos clonales en médula ósea 10-60 %
|
Habitualmente IgG >30 g/l o IgA >20 g/l en suero o >500 mg/24 h en orina
|
Ausente
|
Como en MM
|
|
Infiltrado de plasmocitos en médula ósea
|
<10 %
|
<60 %
|
Habitualmente como en GMSI <10 %
|
<10 %
|
>10 %c
|
>10 %c
|
>10 %c
|
|
>1 de los criterios CRAB-SLiM (→Mieloma múltiple, Criterios diagnósticos)
|
–
|
–d
|
–d
|
–d
|
–
|
–
|
–
|
+
|
+
|
+
|
|
Plasmocitomas
|
–
|
–
|
–
|
–
|
–
|
1 tumor
|
–
|
–/+
|
–/+
|
–/+
|
|
Síndrome nefrótico o albuminuria subnefrótica
|
–
|
+/–
|
+
|
+/–
|
–
|
–
|
–
|
–e
|
–e
|
–e
|
|
Polineuropatía
|
–
|
–/+
|
–/+
|
–/+
|
+
|
–
|
–
|
–e
|
–e
|
–e
|
|
Espleno- o hepatomegalia
|
–
|
–
|
–/+
|
–/+
|
+/–
|
–
|
–
|
–/+
|
–/+
|
–/+
|
|
Edemas, derrame pleural
|
–
|
–/+
|
–/+
|
–/+
|
–/+
|
–
|
–
|
–
|
–
|
–/+
|
|
a También se presentan otras manifestaciones.
b En todas las discrasias pueden presentarse cadenas ligeras libres monoclonales, que se observan en mayores concentraciones en el mieloma de cadenas ligeras.
c Con un porcentaje <10 % es necesario confirmar la presencia de plasmocitoma mediante biopsia.
d Puede presentarse insuficiencia renal por nefropatía po cilindros como en MM.
e Puede presentarse si coexiste AL o EDCL.
AL — amiloidosis de cadenas ligeras de inmunoglobulinas, EDCL — enfermedad por depósito de cadenas ligeras, GMSI — gammapatía monoclonal de significado incierto, GMSR — gammapatía monoclonal de significado renal, MM — mieloma múltiple, MMA — mieloma múltiple asintomático (latente), POEMS — síndrome POEMS (polineuropatía, organomegalia, endocrinopatía, proteína M, lesiones cutáneas)
|
Esquemas utilizados en la primera línea de tratamiento de mieloma múltiple
|
Fármaco
|
Dosis y vía de administración
|
Día de administración
|
Notas
|
|
VTD
|
|
Bortezomib (V)
|
1,3 mg/m2 iv. o VSc
|
1, 4, 8, 11
|
Ciclos repetidos cada 3 semanas
|
|
Talidomida (T)
|
100 mg VO
|
Todos los días
|
|
Dexametasona (D)
|
20 mg VO
|
1, 2, 4, 5, 8, 9, 11, 12
|
|
VD
|
|
Bortezomib
|
1,3 mg/m2 iv. o VSc
|
1, 4, 8, 11
|
Ciclos repetidos cada 3 semanas
|
|
Dexametasona
|
20-40 mg VO
|
1-4, 9-12, 17-20
|
|
VCD
|
|
Bortezomib
|
1,3 mg/m2 iv. o VSc
|
1, 4, 8, 11
|
Ciclos repetidos cada 4 semanas
|
|
Ciclofosfamida
|
300 mg/m2 VO
|
1, 8, 15, 22
|
|
Dexametasona
|
40 mg VO
|
1-4, 9-12, 17-20
|
|
VMP (ciclos repetidos cada 6 semanas, hasta 9 repeticiones)
|
|
Bortezomib
|
1,3 mg/m2
|
iv. o VSc
|
1, 4, 8, 11, 22, 25, 29, 32 en los ciclos 1-4, y en los ciclos 5-9: 1, 8, 22, 29
|
|
Melfalán
|
9 mg/m2
|
VO
|
1-4 en cada ciclo
|
|
Prednisona
|
60 mg/m2
|
VO
|
1-4 en cada ciclo
|
|
RVD
|
|
Lenalidomida
|
25 mg VO
|
1-14
|
Ciclos repetidos cada 3 semanas (ciclos 1-8)
|
|
Bortezomib
|
1,3 mg/m2 iv. o VSc
|
1, 4, 8, 11
|
|
Dexametasona
|
20 mg VO
|
1, 2, 4, 5, 8, 9, 11, 12
|
|
Lenalidomida
|
25 mg VO
|
1-21
|
Ciclos de 28 días
(desde el ciclo 9)
|
|
Bortezomib
|
1,3 mg/m2 iv. o VSc
|
1, 4, 8, 11
|
|
Dexametasona
|
40 mg VO
|
1, 8, 15, 22
|
|
RD
|
|
Lenalidomida
|
25 mg VO
|
1-21
|
Ciclos de 28 días
|
|
Dexametasona
|
40 mg VO
|
1, 8, 15, 22
|
|
dara-VTF
|
|
Daratumumab
|
16 mg/kg iv.
o
1800 mg VSc
|
En el tratamiento de inducción: 1 × semana (las semanas 1-8), cada 2 semanas (las semanas 9-16);
En el tratamiento de consolidación: cada 2 semanas
|
Ciclos repetidos cada 3 semanas
|
|
Bortezomib
|
1,3 mg/m2 iv. o VSc
|
1, 4, 8, 11
|
|
Dexametasona
|
40 mg VO
20 mg VO
|
1, 2, 8, 9, 15, 16, 22, 23 (ciclos 1-2); 1, 2 (ciclos 3-4)
8, 9, 15, 16 del ciclo 3-4
1, 2, 8, 9, 15, 15 del ciclo 5-6
|
|
Talidomida
|
100 mg VO
|
Uso continuo
|
|
Esquemas seleccionados de tratamiento de mieloma múltiple refractario y recidivante
|
Fármaco
|
Dosis y vía de administración
|
Días de administración
|
Notas
|
|
KD
|
|
Carfilzomib
|
20 mg/m2 iv. en el 1.er ciclo
56 mg/m2 iv. en el 1.er ciclo
|
1, 2, 8, 9, 15, 16
|
Ciclos repetidos cada 4 semanas
|
|
56 mg/m2 iv. en los siguientes ciclos
|
1, 2, 8, 9, 15, 16
|
|
Dexametasona
|
20 mg VO o iv.
|
1, 2, 8, 9, 15, 16, 22, 23
|
|
KRD
|
|
Carfilzomib
|
20 mg/m2 iv. en los primeros 2 días del 1.er ciclo
|
1, 2, 8, 9, 15, 16
|
Ciclos repetidos cada 28 días
|
|
27 mg/m2 iv. en los siguientes ciclos
|
1, 2, 8, 9, 15, 16
|
|
Lenalidomida
|
25 mg VO
|
1-21
|
|
Dexametasona
|
40 mg VO
|
1, 8, 15, 22
|
|
IRD
|
|
Ixazomib
|
4 mg VO
|
1, 8, 15
|
Ciclos repetidos cada 4 semanas
|
|
Lenalidomida
|
25 mg/d VO
|
1-21
|
|
Dexametasona
|
40 mg VO
|
1, 8, 15, 22
|
|
|
RD
|
|
Lenalidomida
|
25 mg VO
|
1-21
|
Ciclos de 28 días
|
|
Dexametasona
|
40 mg VO
|
1-4 o 1, 8, 15, 22
|
|
PomDex
|
|
Pomalidomida
|
4 mg VO
|
1-21
|
Ciclos repetidos cada 4 semanas hasta la progresión
|
|
Dexametasona
|
40 mg VO
|
1, 8, 15, 22
|
|
VRD
|
|
Bortezomib
|
1,3 mg/m2 iv.
|
1, 8, 15
|
Ciclos repetidos cada 3 semanas
|
|
Lenalidomida
|
25 mg VO
|
1-14
|
|
Dexametasona
|
40 mg VO
|
1, 8, 15
|
|
DVD
|
|
Daratumumab
|
16 mg/kg iv.
|
Cada semana durante los primeros 3 ciclos
Cada 3 semanas en ciclos 4-8, posteriormente cada 4 semanas
|
Ciclos repetidos cada 4 semanas
|
|
Bortezomib
|
1,3 mg/m2 de sc.
|
1, 4, 8, 11 (ciclos 1-8)
|
|
Dexametasona
|
20 mg VO
|
1-2, 4-5, 8-9, 11-12 (ciclos 1-8)
|
|
EloPd
|
|
Elotuzumab
|
10 mg/kg iv.
20 mg/kg iv.
|
1, 8, 15, 22 (ciclos 1-2)
1 (desde el ciclo 3)
|
Ciclos repetidos cada 4 semanas
|
|
Pomalidomida
|
4 mg VO
|
1-21
|
|
Dexametasona
|
8 mg iv. o 28 mg VO (≤75 años) u 8 mg VO (>75 años)
40 mg VO (≤75 años) o 20 mg VO (>75 años)
|
Los días de administración de elotuzumab
8, 15, 22 (desde el ciclo 3)
|
|
VTD-PACE
|
|
Bortezomib
|
1 mg/m2 de sc.
|
1, 4, 8, 11
|
Ciclos repetidos cada 4 semanas
|
|
Talidomida
|
50-200 mg VO
|
4-7
|
|
Dexametasona
|
40 mg VO
|
4-7
|
|
Cisplatino
|
10 mg/m2 iv.
|
4-7
|
Infusiones continuas de 24 h
|
|
Doxorrubicina
|
10 mg/m2 iv.
|
4-7
|
|
Ciclofosfamida
|
400 mg/m2 iv.
|
4-7
|
|
Etopósido
|
40 mg/m2 iv.
|
4-7
|
Evaluación de la respuesta al tratamiento en mielóma múltiple según la IMWG (2016)
|
Tipo de respuesta
|
Criterios
|
|
Remisión completa estricta (RCs)
|
– cumplidos los criterios de RC (→más adelante) y
– relación normal de sFLC κ/λ, y
– en la inmunohistoquímica de la médula ósea no se detectan plasmocitos monoclonales (relación κ/λ ≤4:1 o ≥1:2 para las cadenas monoclonales κ o λ respectivamente, tras evaluar ≥100 plasmocitos)
|
|
Remisión completa (RC)a
|
– no se detecta proteína M en suero ni en orina mediante la inmunofijación, o
– relación normal de κ/λ si la enfermedad puede medirse solo mediante sFLCb y
– resolución de todos los cambios extramedulares (en tejidos blandos), y
– ≤5 % de plasmocitos en aspirado de médula ósea
|
|
Respuesta parcial muy buena (RPMB)
|
– no se detecta proteína M en electroforesis de suero y de orina (se detecta por inmunofijación) o
– disminución de los niveles de proteína M en suero en ≥90 % y su eliminación por la orina <100 mg/d
– disminución de dFLC en >90 % si la enfermedad puede medirse solo mediante sFLCb
– reducción del tamaño de los eventuales plasmocitomas de tejidos blandos en >90 %c
|
|
Remisión parcial (RP)
|
– disminución de los niveles de proteína M en suero en ≥50 % o su eliminación por la orina en 24 h ≥90 % o hasta <200 mg, o
– disminución de dFLC en ≥50 %, cuando la proteína M no es medible ni en suero ni en orinab, y
– disminución del porcentaje de plasmocitos en médula en ≥50 %, si inicialmente fue ≥30 %, cuando la proteína M no es medible ni en suero ni en orina y no se puede medir sFLCd
– reducción del tamaño de los eventuales plasmocitomas de tejidos blandos en ≥50 %c
|
|
Respuesta mínima (RM)
|
– disminución de los niveles de proteína M en suero en ≥25 %, pero ≤49 %, y disminución de su eliminación por la orina en 24 h en un 50-89 %, y
– reducción del tamaño de los eventuales plasmocitomas de tejidos blandos en un 25-49 %c
|
|
Enfermedad estable (EE)
|
No se recomienda como indicador de respuesta al tratamiento; significa que no se cumplieron los criterios de RC, RPMB, RP, RM ni de enfermedad progresiva
|
|
Enfermedad progresiva (EP)
|
≥1 de los siguientes:
– aumento de la proteína M en suero en >25 %, con un aumento absoluto en ≥5 g/l
– aumento de la proteína M en suero en ≥10 g/l, con una concentración inicial ≥5 g/l
– aumento de la eliminación de la proteína M por la orina en ≥25 %, con un aumento absoluto en ≥200 mg/d
– aumento de dFLC en ≥25 %, con un aumento absoluto en >100 mg/l en enfermos cuando la proteína M no es medible ni en suero ni en orinab
– aumento del porcentaje de plasmocitos en médula ósea en ≥25 % en relación al porcentaje inicial, con un aumento absoluto de este porcentaje en >10 % cuando la proteína M no es medible ni en suero ni en orina y no se puede medir sFLCd
– aparición o aumento de tamaño de lesiones óseas o de tumores extramedulares en tejidos blandos (en ≥50 % de la suma de los productos de las dimensiones perpendiculares de >1 lesión, o en ≥50 % de la dimensión longitudinal de una lesión de >1 cm en eje corto)c
– aumento en ≥50 % del recuento de plasmocitos circulantes (mín. 200 células/µl), si es la única manera de evaluación de la enfermedad
|
|
Recidiva clínica
|
≥1 de los siguientes:
– criterio de afectación orgánica relacionado con el MM (CRAB)
– aparición de nuevos plasmocitomas de tejidos blandos o de nuevas lesiones óseas (excepto fracturas por osteoporosis)
– aumento del diámetro de los plasmocitomas o de las lesiones óseas ya existentes en un 50 % y ≥1 cmc
– hipercalcemia >11 mg/dl
– disminución de los niveles de hemoglobina en ≥2 g/dl no asociada al tratamiento ni a causas distintas al mieloma
– síndrome de hiperviscosidad debido a la presencia de paraproteína en suero
|
|
Si se cumplen los criterios de cualquier categoría de respuesta, es necesario realizar 2 mediciones más antes de introducir una nueva línea de tratamiento (pueden ser 2 muestras distintas tomadas el mismo día).
Además, para confirmar cualquier tipo de respuesta al tratamiento es necesario que no se presenten nuevas lesiones óseas o plasmocitomas extramedulares, y que los ya existentes no aumenten de tamaño (si se han realizado estudios radiológicos).
sFLC sirven para evaluar la respuesta solo cuando la proteína M no es medible ni en suero ni en orina. Criterios de enfermedad medible: proteína M en suero ≥10 g/l, proteína M en orina ≥200 mg/24 h, sFLC clonal ≥100 mg/l (siempre y cuando la relación κ/λ sea anormal).
En caso de mieloma IgA e IgD, para evaluar la respuesta es preferible la determinación de las concentraciones de la inmunoglobulina correspondiente en vez del pico de la proteína M en la electroforesis.
En mieloma biclonal, para evaluar la respuesta se suman las concentraciones de ambos picos de la proteína M.
En algunos enfermos con mieloma después del tratamiento, sobre todo en RC tras auto-PBSCT, se desarrolla la denominada GMSI secundaria, es decir la presencia de proteína monoclonal de un isotipo distinto al inicial en concentraciones bajas (inmunofijación). Es un fenómeno transitorio (significa una reconstitución clonal del sistema inmunológico), no significa progresión de la enfermedad y no requiere tratamiento.
El uso de anticuerpos monoclonales (p. ej. daratumumab) puede causar la aparición de IgG κ monoclonal en inmunofijación de suero.
a Además, en los ensayos clínicos en el marco de RC se evalúa la presencia de enfermedad mínima residual (EMR) en médula mediante citometría de flujo de nueva generación (next-generation flow, NGF) o mediante secuenciación del ADN de nueva generación (next-generation sequencing, NGS), y eventualmente con PET-TC.
b Se refiere al mieloma de cadenas ligeras y a algunos casos de mieloma no secretor.
c El tamaño de los plasmocitomas de tejidos blandos (extramedulares) se expresa como suma de productos de las dimensiones perpendiculares de las lesiones en pruebas de imagen (TC, PET-TC, RMN), o se mide con regla en caso de lesiones cutáneas. En la evaluación de la respuesta no se toman en cuenta los tumores tras radioterapia, aunque requieren vigilancia para detectar una eventual EP.
d Se refiere al mieloma no productor y a algunos casos de mieloma no secretor.
e Un resultado positivo de inmunofijación aislado en caso de RC anterior no significa EP.
dFLC — diferencia entre la concentración de sFLC clonales y sFLC no asociadas a mieloma (normales), RC — respuesta completa, sFLC — cadenas ligeras libres en suero
|
Dosis profilácticas de HBPM en enfermos no operados y en embarazadas
|
HBPMa
|
Dosis profilácticas
|
|
Enfermos no operados
|
Embarazadasb
|
|
Dalteparina
|
5000 UI cada 24 h
|
5000 UI VSc cada 24 h
|
|
Enoxaparina
|
40 mg cada 24 h
|
40 mg VSc cada 24 hc
|
|
Nadroparina
|
2850 UI cada 24 h
|
3800 UI VSc cada 24 h
|
|
a Preparados →tabla 2 en Trombosis venosa profunda.
b Puede ser necesaria una modificación de la dosis en el 3.er trimestre (según la valoración de la actividad anti-Xa, actividad objetivo 0,2-0,5 UI/ml 3-4 h después de la inyección VSc).
c Puede ser necesaria una modificación de la dosis en caso de peso corporal extremadamente bajo o alto.
HBPM — heparina de bajo peso molecular
|
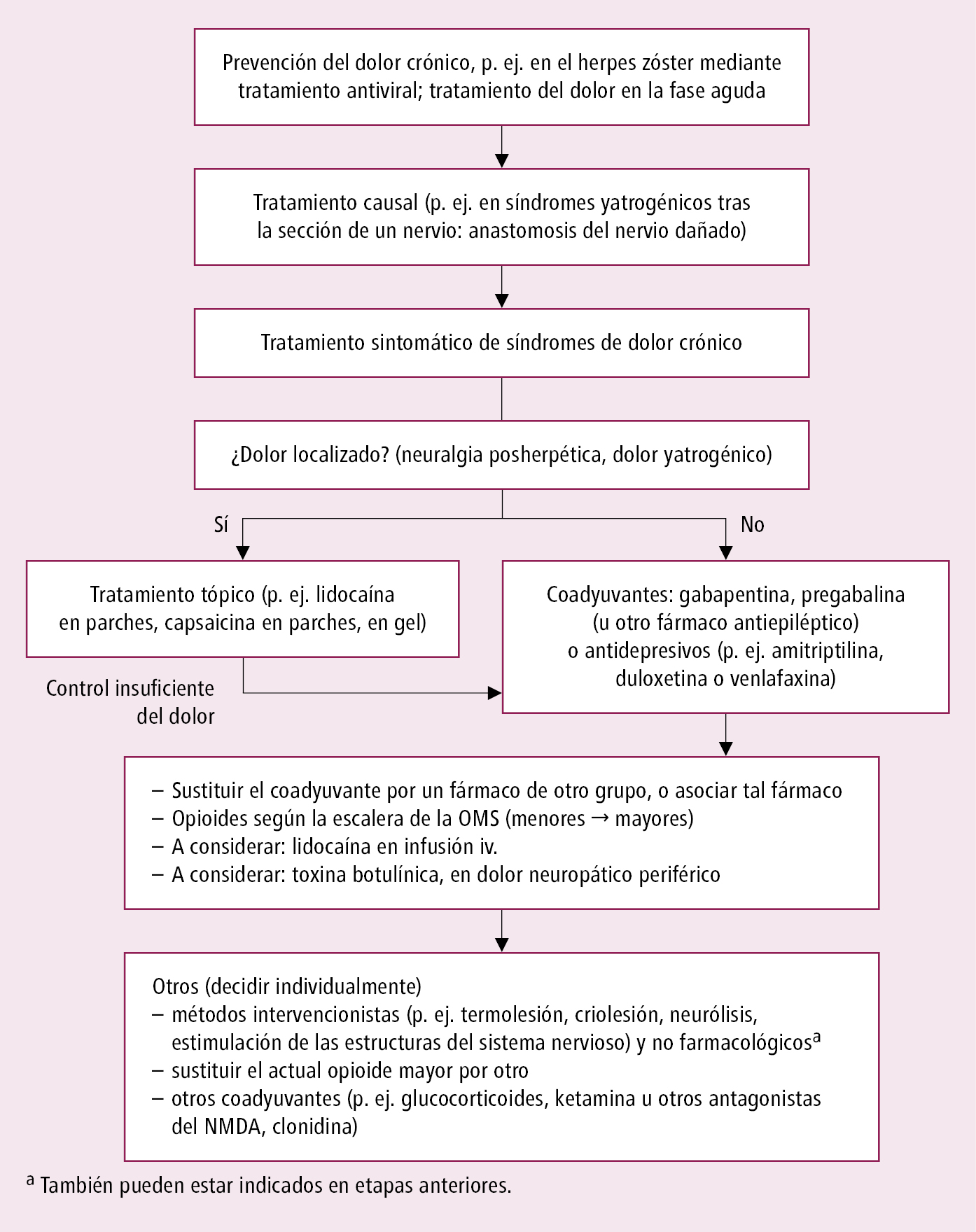
Fig. 24.1-2. Algoritmo de tratamiento del dolor neuropático puro
 Español
Español
 English
English
 українська
українська

